Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(1):189-203. doi:10.7150/ijbs.97886 This issue Cite
Research Paper
METTL16 suppresses ferroptosis in cholangiocarcinoma by promoting ATF4 via m6A modification
Senfeng Zhao1,2,3,#, Jiahui Cao1,2,3,#, Ruopeng Liang1,2,3,#, Tingting Peng4, Shitao Wu1,2,3, Zhipu Liu1,2,3, Yahui Wu1,2,3, Liming Song1,2,3,5, Chenguang Sun1,2,3, Yin Liu1,2,3, Junmou Gu1,2,3, Libo Wang1,2,3, Rongtao Zhu1,2,3, Weijie Wang1,2,3, Yuling Sun1,2,3, ![]()
1. Department of Hepatobiliary and Pancreatic Surgery, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China.
2. Institute of Hepatobiliary and Pancreatic Diseases, Zhengzhou University, Zhengzhou, China.
3. Zhengzhou Basic and Clinical Key Laboratory of Hepatopancreatobiliary Diseases, Zhengzhou, China.
4. Department of Neurology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China.
5. Department of Hepatobiliary and Pancreatic Surgery, Zhengzhou Central Hospital Affiliated to Zhengzhou University, Zhengzhou, China.
# These authors contributed equally: Senfeng Zhao, Jiahui Cao, Ruopeng Liang.
Received 2024-4-29; Accepted 2024-11-8; Published 2025-1-1
Abstract

Background: N6-methyladenosine (m6A) modification is the most common post-transcriptional modifications, which is critical for the metabolism of ferroptosis-related RNAs. Yet, the impact of m6A modification on ferroptosis in cholangiocarcinoma (CC) is far from clear.
Methods: Public databases and tissue arrays were applied to explore the clinical relevance of METTL16 in CC. Then, the effects of METTL16 on growth and ferroptosis were studied in vitro and in vivo. Mechanistically, RNA-sequencing, methylated RNA immunoprecipitation, dual-luciferase reporter assays and RNA stability assays were used to identify the METTL16/ATF4 axis in ferroptosis in CC.
Results: Clinically, we find that METTL16 is overexpressed and associated with a poor prognosis in patients with CC. Functionally, METTL16 protects against ferroptosis by maintaining mitochondrial homeostasis, including mitochondrial structure, membrane potential and energy products. It also decreases cellular metabolism of Fe2+ and lipid peroxide, thereby promoting cell growth in vitro and in vivo. Mechanistically, ATF4 is a novel target of METTL16 and METTL16 enhances the m6A level and expression of ATF4 mRNA by inhibiting its decay, which further prevented ferroptosis in CC via m6A modification.
Conclusions: Our findings highlighted the role of METTL16/ATF4 in ferroptosis, which sheds light on potential therapeutic strategies for CC.
Keywords: m6A, ferroptosis, METTL16, ATF4, cholangiocarcinoma
Introduction
Cholangiocarcinoma (CC) is a rare malignancy, accounting for less than 3% of gastrointestinal tumors, but its morbidity and mortality have been increasing worldwide in recent decades [1-3]. Currently, surgery remains the preferred treatment for early-stage CC, and the outcomes doesn't come into satisfaction because of high tumor aggressiveness and recurrence. Despite the application of targeted agents combined with checkpoint inhibitors, advanced CC remains problematic due to drug resistance and high tumor heterogeneity [4, 5]. Therefore, it is imperative to deepen the understand underlying occurrence and development of CC to improve future treatment outcomes.
N6-methyladenosine (m6A), which was initially identified in 1974, is the predominant post-transcriptional modification of RNA. It is involved in almost full processes of RNA metabolism, such as RNA processing, transport, translation and decay [6-8]. To date, three classes of enzymes have been shown to keep this modification reversible and dynamic: methyltransferases, demethylases and reading proteins. Methyltransferases, which are exemplified by METTL3, METTL14, METTL16 and WTAP, cause RNA methylation. In contrast, demethylases including ALKBH5 and FTO remove methylation from RNA. In addition, m6A motifs are recognized and bound by reading proteins, such as YTHDF/2/3, IGF2BP1/2/3 and YTHDC1/2 [8, 9]. Previous researches have revealed that m6A modification have great effect on various cellular processes, including proliferation, differentiation and death [10, 11]. Intriguingly, resistance to cell death is a characteristic hallmark of tumors, facilitating cancer progression and acquired resistance to anti-tumor therapies [12]. However, as comparatively few studies on this topic, less is known about m6A in cell death of CC.
Ferroptosis is a unique programmed cell death distinguished by the accumulation of Fe2+ and lipid peroxidation (LPO) [13]. Morphologically, ferroptosis is characterized by mitochondrial shrinkage, with a decrease or vanishing of mitochondrial cristae and a rise in mitochondrial membrane density [14, 15]. Increasing studies have highlighted the the vital importance of ferroptosis in the tumorigenesis of cancers, such as colon cancer, live cancer, and pancreatic cancer [16-18]. Therefore, novel pharmacological strategies targeting ferroptosis have emerged as therapeutic interventions. Agents like sorafenib and sulfasalazine have been used in the treatment of hepatocellular carcinoma by inducing ferroptosis [19, 20]. Furthermore, ATF4 plays a vital role in this process. ATF4, a transcription factor, activates SLC7A11 during oxidative stress, contributing to the transport of extracellular cystine and the production of glutathione (GSH), thereby preventing ferroptosis [21, 22]. It also promotes the expression of HSPA5, which binds to GPX4 to arrest its degradation and subsequent lipid peroxidation, further protecting against ferroptosis [23]. Nevertheless, ATF4-mediated ferroptosis in CC remains incompletely understood.
Here, we analyzed the expression of m6A-associated genes in CC and explored the underlying mechanisms. Clinically, the m6A methyltransferase METTL16 was upregulated and acted as an independent prognostic factor of CC. Mechanistically, METTL16 regulated the migration and proliferation of CC by suppressing ferroptosis through ATF4 in an m6A-dependent manner. In total, we highlighted the importance of METTL16-mediated m6A modification and ferroptosis in CC and provided a promising strategy for the treatment of CC.
Materials and Methods
Cell culture and transfection
The human extrahepatic CC cell line KMBC and intrahepatic CC cell line RBE were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, China), and cultured in RPMI-1640 (Gibco, Invitrogen, Carlsbad, USA) supplemented with 10% fetal bovine serum (Life-iLab, Shanghai, China). siRNAs (si METTL16, si ATF4 and paired control) came from RiboBio (Guangzhou, China) and the sequences showed in Table S1. Plasmids (sh METTL16, METTL16, ATF4 and paired control) came from GenePharma (Shanghai, China). LipofectamineTM 2000 (Invitrogen, Carlsbad, USA) was used for transient transfection.
Cell counting kit-8 (CCK8) assay
As to intervention by erastin (MCE, Shanghai, China) or RSL3 (MCE, Shanghai, China), 5 × 103 normal cells were cultured in 96-well plates overnight. Then, cells were treated with RSL3 or erastin for 24 h and added 10 μL CCK8 reagent (TargetMol, Shanghai, China). As to intervention by liproxstatin or ferrostatin-1, 5 × 103 transfected cells were cultured in 96-well plates overnight. Then, cells were treated with liproxstatin (MCE, Shanghai, China) or ferrostatin-1 (MCE, Shanghai, China) and added 10 μL CCK8 reagent at 12-hour intervals until 48 hours. After incubation for 2 h, the cell viability was measured.
EdU and colony formation assays
The EdU and colony formation assays were used to assess cell proliferative capacity. As to EdU assay, 1 ×104 transfected cells were plated in 24-well plates and cultured overnight. The proportion of proliferative cells was evaluated by EdU Cell Proliferation Image Kit (Abbkine, Wuhan, China), based on images captured by fluorescence microscopy. For colony formation, 1500 transfected cells were cultured in 6-well plates for 2 weeks. The colonies were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet (Solarbio, Beijing, China).
Transwell and wound-healing assays
The Transwell and wound-healing assays were applied to evaluate cell migration ability. For Transwell assay, the upper chambers were cultured with 2 × 104 transfected cells in serum-free medium, and the chambers below were filled with 20% FBS medium. 24 h later, the upper chambers were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. For wound-healing assay, 2.0 × 105 transfected cells were cultured in 12-well plates until to complete confluence. Finally, a scratch was made, and the healing situation was recorded after 0 and 24 h.
Apoptosis
Annexin V/PI apoptosis kit (UElandy, Suzhou, China) were used to assess the cell apoptosis. Briefly, cells in 6-cm dishes were transfected for 48h. According to the manufacturer's instructions, cells were resuspended in staining buffer, and tested by flow cytometry.
Western blotting (WB)
Proteins were obtained from tissues and cells by RIPA buffer (Solarbio, Beijing, China) with protease inhibitors (Beyotime, Shanghai, China). The total protein concentration was determined using a BCA protein assay kit (EpiZyme, Shanghai, China), 20 μg protein were separated by SDS-PAGE and transferred to 0.45 μm PVDF membranes (Millipore, Billerica, USA). Then, the membranes were blocked with 5% non-fat milk at room temperature for 2 h and incubated with primary antibodies at 4°C overnight. Next day, the membranes were incubated with secondary antibodies specific to the primary antibodies at room temperature for 1 h. The visualization of the bound antibodies was achieved using an ECL (Enhanced Chemiluminescence) reagent (Millipore, Billerica, USA). The primary antibodies and secondary antibodies used are detailed in Table S2.
Quantitative reverse transcription PCR (qPCR)
Total RNA was extracted from tissues and cells by TRIzol (CWBIO, Beijing, China), followed by cDNA synthesis with SweScript All-in-One RT SuperMix (Servicebio, Wuhan, China). Subsequently, qPCR was conducted with SYBR Green qPCR Master Mix (Servicebio, Wuhan, China). The specific primers employed for qPCR are detailed in Table S3.
RNA-sequencing
RNA-sequencing services were provided by Kaitaibio (Hangzhou, China). mRNA was enriched from total RNA, and an RNA library was constructed. After the library passed the quality inspection, RNA-sequencing was conducted on an Illumina NovaSeq 6000. The raw data were normalized and analyzed by bioinformatics.
Methylated RNA immunoprecipitation (MeRIP)
Total RNA was obtained by TRIzol, and RNA immunoprecipitation assays were conducted using a Magna RIP Kit (Millipore, Darmstadt, Germany). Using an anti-m6A antibody and normal rabbit IgG to immunoprecipitated RNA-binding proteins. The enriched RNA was released, purified, and eluted by RNA Clean & Concentrator TM (Zymo Research, Irvine, USA). qPCR was performed following MeRIP to quantify changes in the m6A methylation level of target genes. Furthermore, nucleic acid electrophoresis was used to verify the target gene in cDNA from MeRIP-qPCR.
Dual-luciferase reporter assay
ATF4 mRNA fragments (CDS, from 341-488) containing wild-type and mutant m6A motifs (the second predicted site, A was replaced by C) were directly synthesized and used to construct luciferase reporter plasmids by GeneChem (Shanghai, China). Wild-type or mutant ATF4 luciferase plasmids were co-transfected with siMETTL16 and controls. Luciferase activity was quantified by a dual-luciferase reporter assay system (Vazyme, Nanjing, China). The relative activity of the firefly luciferase was standardized by referencing it against the activity of renilla luciferase.
RNA stability assay
For RNA stability assays, cells were transfected with si METTL16, METTL16 plasmids and controls. Then, the cells were exposed to 10 μg/ml actinomycin D (Med Chem Express, New Jersey, USA) for 0 h, 2 h, 4 h, and 8 h. Subsequently, total RNA was extracted, and the ATF4 mRNA levels were quantified by qPCR.
Superoxide dismutase (SOD), and antioxidant capacity assays
To determine the SOD and antioxidant capacity levels in cells, Total Superoxide Dismutase Assay Kit with WST-8 (Beyotime, Shanghai, China) and Total antioxidant capacity assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) were used, respectively. Briefly, cells in 6-cm dishes were processed following the manufacturer's protocol, and the absorbance of the reaction mixture was measured at 450 nm and 405 nm, respectively.
GSH, malondialdehyde (MDA) and ATP assays
To measure GSH, MDA and ATP levels in cells, GSH assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China), Lipid Peroxidation MDA Assay Kit (Beyotime, Shanghai, China) and ATP assay kit (Beyotime, Shanghai, China) were used, respectively. In short, cells in 10-cm dishes or tumor tissues from xenografts were lysed with Cell lysis buffer for Western and IP (Beyotime, Shanghai, China) and subsequently analyzed following the manufacturer's instruction. Finally, the absorbance at 532 or 420 nm and the fluorescence intensity were measured.
Mitochondrial membrane potential (MMP), reactive oxygen species (ROS), superoxide anion (SOA), Fe2+ and LPO assays
TMRE (Beyotime, Shanghai, China), DCFH-DA (Beyotime, Shanghai, China), Dihydroethidium (Beyotime, Shanghai, China), Phen Green SK (GlpBio, Montclair, CA, USA) and BODIPY™ 581/591 C11 (Thermo Fisher Scientific, Waltham, USA) were used to assess the MMP, ROS, SOA, Fe2+ and LPO levels in cells, respectively. Briefly, cells in 6-cm dishes were incubated with 10 μM TMRE, 10 μM DCFH-DA, 5 μM Dihydroethidium, 5 μM Phen Green SK and 10 μM BODIPY 581/591 C11 for 30 min. Finally, fluorescence microscopy was used to analyze the MMP, Fe2+, ROS and LPO levels in cells, and flow cytometry were used to analyze the SOA and LPO levels.
Transmission electron microscopy (TEM)
Cells were fixed using 2.5% glutaraldehyde in 0.1 mM phosphate buffer, and incubated in 1% OsO4 for 2 h. Once dehydrated, the cells were embedded in epoxy resin. After resin blocks trimmed, Ultrathin sections were made using an ultramicrotome and stained with lead citrate and uranyl acetate. Finally, the stained ultrathin sections were observed using TEM.
Confocal laser scanning microscopy (CLSM)
CLSM was employed to confirm the location of METTL16 in cells. The cells were fixed with 4% paraformaldehyde for 45 min, permeabilized using 0.3% Triton X-100 for 30 min, and blocked with 10% goat serum for 1 h. Next, the cells were incubated overnight at 4°C with an anti-METTL16 antibody, followed by incubation with goat anti-rabbit IgG fluorescent antibody for 1 h at 37°C. To visualize the cell nuclei, the cells were stained with DAPI (Solarbio, Beijing, China). Finally, the cells were examined using a Zeiss LSM880 CLSM with a 100× oil immersion objective.
Animal models
Animal experiments were approved by Animal Ethics Committee of Zhengzhou University. For xenograft models, female BALB/c nude mice (5 weeks, Beijing HFK Bioscience Co., Ltd., Beijing, China) were used to confirm tumor growth, metastasis and ferroptosis in vivo. KMBC cells (1 × 107 cells) were percutaneously injected into the axilla of mice. After two weeks, the tumors were dissected and divided into 50 cm3 masses. Then, the tumor masses were subcutaneously implanted into the axilla of mice and subsequently injected with sh METTL16, METTL16 and control plasmids along with Lipo8000TM Transfection Reagent (Beyotime, Shanghai, China) every three days to establish METTL16-knockdown and METTL16-overexpressing subcutaneous xenografts. The body weight of the mice and the size of the tumors were measured every three days until the mice were sacrificed. Dissected tumor tissues were preserved at -80°C or 10% formalin fixation and used for later experiments.
Immunohistochemistry (IHC) and hematoxylin eosin (HE) staining
Xenografts were embedded in paraffin and further precisely sliced into 4-μm-thick sections for HE and IHC staining. For HE, the sections were counterstained with hematoxylin (Servicebio, Wuhan, China), followed by incubation in DAB substrate solution (Servicebio, Wuhan, Chinas). For IHC, sodium citrate buffer was used for antigen retrieval. Then, the sections were incubated with 3% hydrogen peroxide for 20 min and blocked with 3% BSA for 1 h at room temperature. Then, the sections were incubated with primary antibodies overnight at 4°C. Following this, a secondary antibody was added and incubated for 60 min at room temperature. All the sections were observed by microscopy.
Bioinformatics analysis
Three gene expression profiles, namely, GSE132305 (extrahepatic CC), GSE107943 (intrahepatic CC) and GSE76297 (only use intrahepatic CC) were obtained from the GEO. Differentially expressed genes were screened with the Limma package. GO and KEGG were used for functional enrichment analysis. The expression of METTL16 was analyzed by DepMap. The proteins level of METTL16 and ATF4 was studied by the Human Protein Atlas (HPA). SRAMP was used to predict m6A modification sites on ATF4 mRNA.
Statistical analysis
All the experiments were independently conducted three times, and the data were analyzed by GraphPad Prism 8. The results are presented as the mean ± standard deviation (SD). The t tests or two-way ANOVA was used to analyze differences in normally distributed data between two groups. The Kaplan-Meier method was employed for survival analysis. Pearson's correlation test was used to assess the correlation between METTL16 expression and ATF4 expression. p<0.05 was considered significant.
Results
METTL16 is overexpressed in CC and serves as a poor prognostic factor
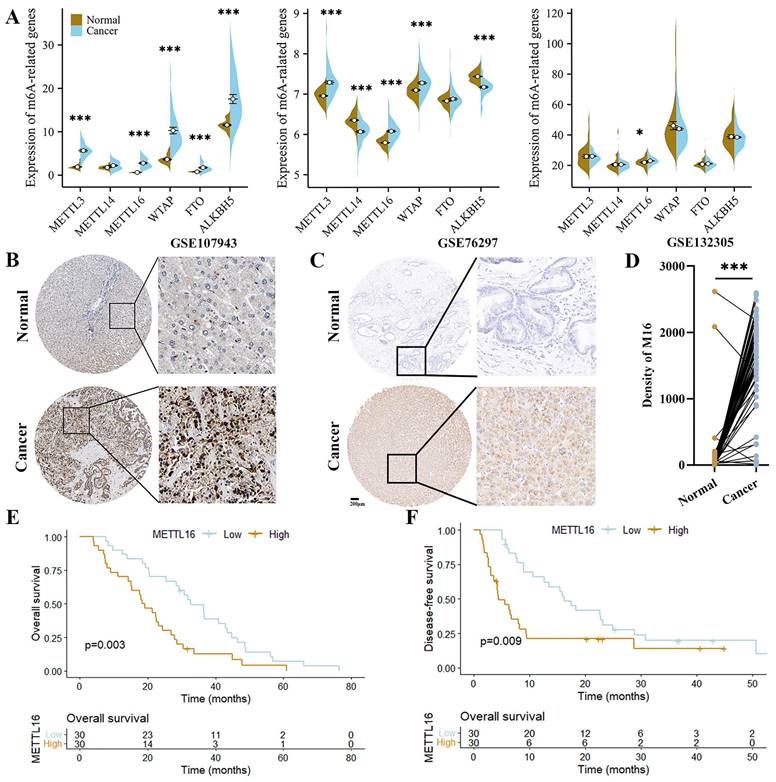
Initially, we queried the expression of the most important m6A regulatory genes in GEO datasets to explore the role of m6A modifications in CC. As shown, METTL3, METTL16 and WTAP were significantly upregulated in CC tissues (2/3 datasets), implying a vital effect of m6A modification on the progression of CC. Among these genes, only METTL16 was elevated in all datasets (Figure 1A). Subsequently, the clinical value of METTL16 was further analyzed with HPA and CC tissue array (60 CC tissues and paired adjacent normal tissues). We found that METTL16 expression was significantly higher in the cancer tissues (Figure 1B-D). Moreover, high METTL16 expression shorten the overall survival (OS) and disease-free survival (DFS) of CC patients (Figure 1E and F). Collectively, these findings suggest that METTL16 is overexpressed in CC and acts a risk factor for CC patient prognosis.
METTL16 (M16) is upregulated in CC. (A) The expression of m6A-related genes from GSE107943, GSE76297 and GSE132305. (B) The images of IHC staining for M16 in CC and normal tissues from HPA. (C-D) The protein level of M16 in the tissue array of CC was examined by IHC (n=60). (E-F) Kaplan-Meier analysis of the OS (E) and DFS (F) of CC patients from the tissue array. The data are presented as the mean ± SD. * p < 0.05, *** p < 0.005.
METTL16 enhances tumorigenicity in CC
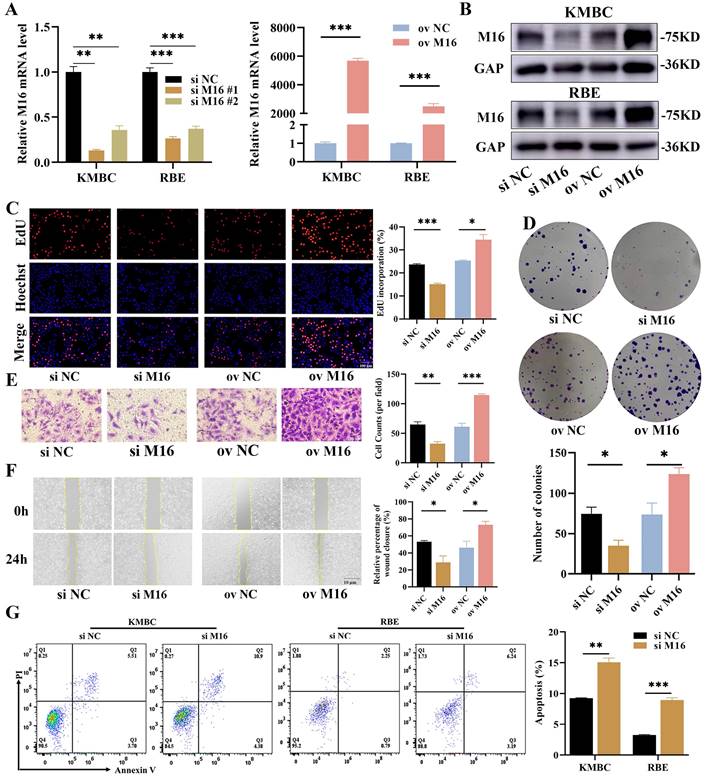
Given that METTL16 is highly expressed in CC, we speculated that METTL16 may have pro-cancer effects in CC. We analyzed the the expression of METTL16 in CC cell lines by DepMap (Figure S1A), and constructed METLL16-knockdown and METLL16-overexpressing cells in KMBC and RBE by siRNAs and METTL16 overexpression plasmid to define the functions of METTL16 in CC (Figure 2A and B). CCK8 assay was used and showed that the survival was reduced after METTL16 knockdown (Figure S1B, #1 sequence of METTL16 was used after). We performed proliferative assays and found that proliferation ratio was much lower in METTL16-knockdown cells, while that was higher in METTL16-overexpressing cells (Figure 2C and D, Figure S1C and D). Then, Transwell and wound-healing assays were used to evaluate the cell migration and the results displayed that METTL16-knockdown cells exhibited slower migration, while METTL16-overexpressing cells exhibited the opposite trend (Figure 2E and F, Figure S1E and F). Next, the cell apoptosis assays were applied and showed that there is a lower apoptotic in METTL16-knockdown cells (Figure 2G). Overall, these results indicate that METTL16 promotes growth in CC in vitro.
M16 promotes CC progression. (A-B) M16 knockdown or overexpression in KMBC and RBE cells was confirmed by qPCR (A) and WB (B). (C-F) The proliferation and migration of M16-knockdown or M16-overexpressing KMBC cells were investigated by EdU assays (C), colony formation assays (D), Transwell assays (E) and wound-healing assays (F). (G) the apoptosis in M16-knockdown KMBC and RBE cells were measured by flow cytometry. The data are presented as the mean ± SD. GAP: GAPDH. * p < 0.05, ** p < 0.01, *** p < 0.005.
METTL16 inhibits oxidative stress in CC
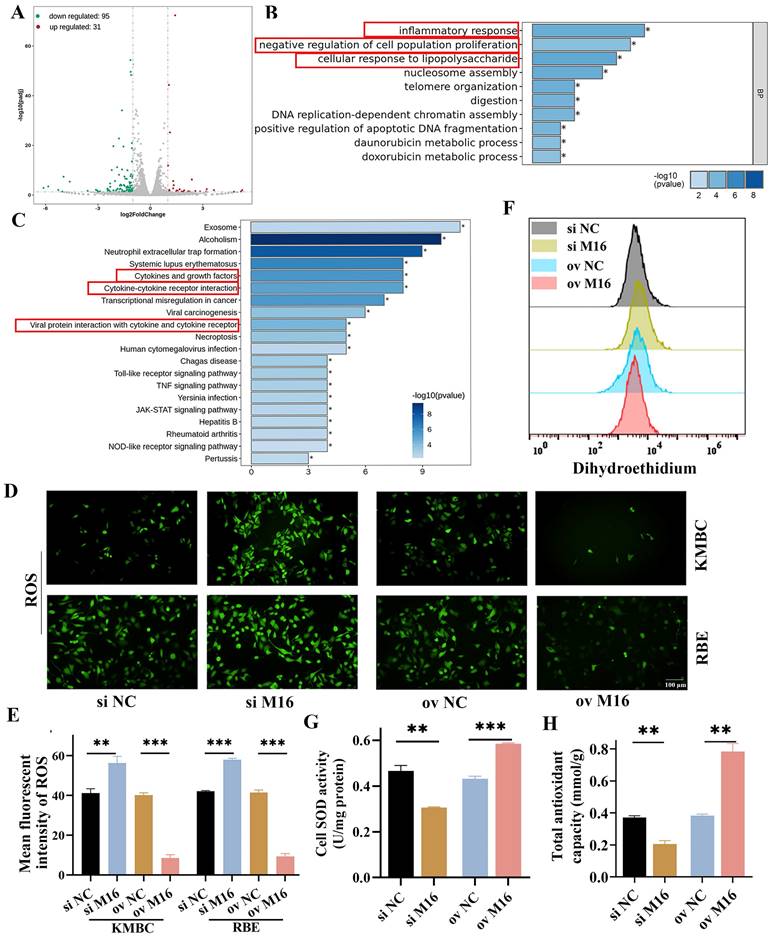
The results described above demonstrated that METTL16 is essential for the development of CC. However, the exact mechanism involved in this process remains unclear. To comprehensively explore the mechanism by which METTL16 regulates CC, RNA-sequencing was performed. We found 95 downregulated genes and 31 upregulated genes in METTL16-knockdown KMBC cells relative to control cells (Figure 3A). Then, these differential genes were analyzed by GO and KEGG and indicated that METTL16 has important effects on the oxidative stress and cell proliferation, such as the inflammatory response, cellular response to cytokine‒cytokine receptor interaction, lipopolysaccharide, negative regulation of cell population proliferation, cytokines and growth factors, and viral protein interaction with cytokines and cytokine receptors (Figure 3B and C). SOD, SOA, ROS and antioxidant capacity levels were main indicators of oxidative stress [24-27]. Hence, we measured intracellular ROS, SOA, SOD and antioxidant capacity levels in KMBC or RBE cells. The results showed that METTL16 knockdown increased intracellular ROS and SOA but reduced SOD and antioxidant capacity levels, whereas METTL16 overexpression decreased intracellular ROS and SOA but elevated SOD and antioxidant capacity levels (Figure 3D-H). In sum, these findings show that METTL16 regulates CC through oxidative stress.
M16 is closely related to oxidative stress. (A) Volcano plot showing differential genes (|log2(fold change)|> 1 and p < 0.05) between M16-knockdown and control KMBC cells. (B-C) GO (B) and KEGG (C) analysis indicated that oxidative stress and cell proliferation were largely affected by M16 knockdown. (D-E) ROS levels in M16-knockdown or M16-overexpressing KMBC and RBE cells were assayed by fluorescence microscopy. (F) SOA levels in M16-knockdown or M16-overexpressing KMBC cells were assayed by flow cytometry. (G-H) SOD (G), total antioxidant capacity (H) levels in M16-knockdown or M16-overexpressing KMBC cells were measured. The data are presented as the mean ± SD. ** p < 0.01, ** p < 0.01, *** p < 0.005.
METTTL16 inhibits ferroptosis in CC
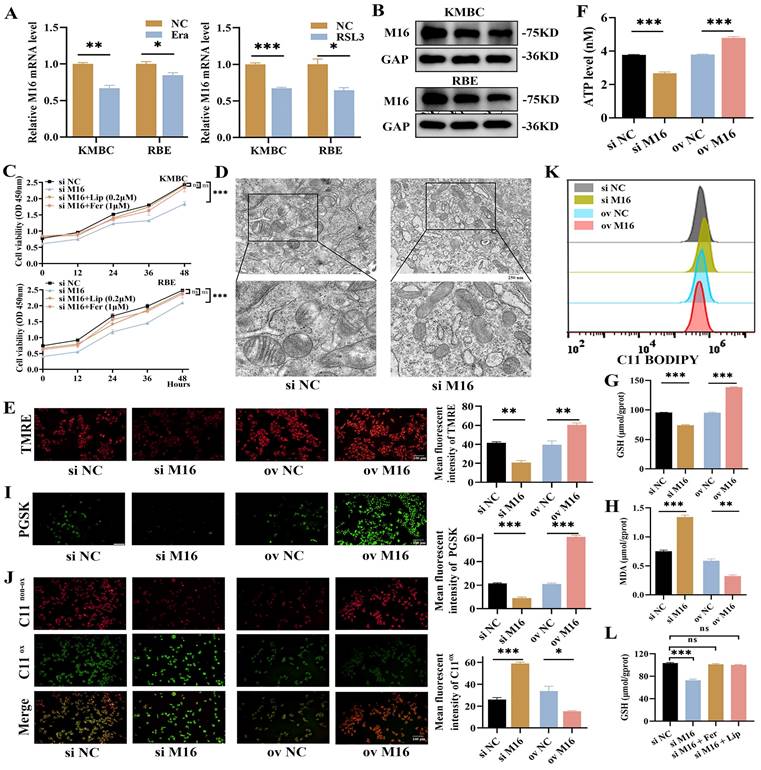
Given that the process of ferroptosis intricately involves oxidative stress, we gave more insight to the link between METTL16 and ferroptosis. We employed RSL3 and erastin to elicit ferroptosis in CC cells (Figure S2A) and observed that METTL16 was significantly decreased during this process (Figure 4A and B), meaning that METTL16 is related to ferroptosis. Furthermore, we employed liproxstatin and ferrostatin-1 to inhibit ferroptosis in KMBC and RBE cells and found that METTL16 knockdown slows cell proliferation which is rescued by liproxstatin or ferrostatin-1 (Figure 4C), suggesting that METTL16 knockdown promotes ferroptosis in CC. Morphologically, ferroptosis has characteristic pathological changes, such as shrinkage of mitochondria, decrease or vanishing of mitochondrial cristae [14]. Then, we observed changes in subcellular organelles in METTL16-knockdown KMBC and RBE cells by TEM and found that mitochondria became smaller and cristae decreased in METTL16-knockdown cells (Figure 4D, Figure S2B). In light of the fact that MMP and intracellular ATP produced mainly by mitochondria are closely related to mitochondrial cristae, the MMP and ATP were measured. The results showed that METTL16 knockdown decreased MMP and ATP, while METTL16 overexpression increased MMP and ATP (Figure 4E and F, Figure S2C). These findings verified that METTL16 knockdown elicits ferroptosis in CC. Biochemically, ferroptosis results from Fe2+ accumulation and LPO. Next, intracellular Fe2+ and metabolic product of LPO, such as GSH, MDA and LPO, were examined [28-30]. The results showed that METTL16 knockdown decreased intracellular GSH levels but increased intracellular MDA, Fe2+ and LPO levels in KMBC and RBE cells. Conversely, METTL16 overexpression elevated intracellular GSH levels but decreased intracellular MDA, Fe2+ and LPO levels (Figure 4G-K, Figure S2D-H). In addition, the effects of METTL16 knockdown on GSH was revised by liproxstatin and ferrostatin-1 in KMBC cells (Figure 4L). In conclusion, these observations indicate that METTL16 suppresses ferroptosis in CC.
M16 inhibits ferroptosis in CC. (A-B) The expression of M16 was assayed in KMBC and RBE cells treated with erastin (20 μM) or RSL3 (1 μM) for 24 h by qPCR (A) and WB (B). (C) The proliferative effects of combination of knockdown M16 and liproxstatin or ferrostatin-1 in KMBC and RBE cells. (D-F) Mitochondria morphology (D), mitochondrial membrane potential (E) and intracellular ATP (F) were observed in KMBC cells transfected with si NC or si M16. (G-I) GSH (G), MDA (H), and Fe2+ levels (I) in M16-knockdown or M16-overexpressing KMBC cells were measured. (J-K) Lipid peroxide levels in M16-knockdown or M16-overexpressing KMBC cells were assayed by fluorescence microscopy (J) and flow cytometry (K) using C11-BODIPY. (L) The GSH effects of combination of knockdown M16 and liproxstatin (0.2 μM) and ferrostatin-1 (1 μM) in KMBC cells. The data are presented as the mean ± SD. * p < 0.05, ** p < 0.01, *** p < 0.005.
ATF4 is a key target of METTL16 and its stability is regulated by METTL16 via m6A modification
Previous studies have shown that ferroptosis is regulated by several key genes, including NCOA4, ATF4, GPX4, SLC7A11 and NRF2. Among these genes, NCOA4 promotes ferroptosis, while the others inhibit ferroptosis [14]. To detail the mechanism in ferroptosis regulated by METTL16, we reviewed the RNA-sequencing results and discovered that METTL16 knockdown reduced the expression of ATF4, NCOA4 and NRF2 (Figure 5A). Afterward, we further verified the impacts of METTL16 on these genes when knockdown and overexpression METTL16 in KMBC and RBE cells. In KMBC cells, METTL16 knockdown decreased the NCOA4 and ATF4 but elevated the SLC7A11, and METTL16 overexpression elevated the ATF4 and NCOA4 but reduced the SLC7A11 (Figure 5B). In RBE cells, METTL16 knockdown reduced the ATF4 and NRF2 but elevated SLC7A11, and METTL16 overexpression increased the ATF4, NCOA4 and NRF2 (Figure S3A). These results showed that only ATF4 is consistently influenced by METTL16. Furthermore, WB revealed that METTL16 knockdown reduced ATF4 which was elevated after METTL16 overexpression in vitro (Figure 5C). In parallel, our analysis of GSE107943, GSE76297 and GSE132305 revealed a positive correlation between METTL16 and ATF4 (Figure S3B). We also knockdown other key m6A-reated genes, like METTL3, METTL14, METTL16, WTAP, ALKBH5, FTO, and found that other genes did not produce similar significant effects on ATF4 mRNA as METTL16 (Figure S3C). Thus, these findings confirm that ATF4 is a major regulatory target of METTL16.
M16 enhances ATF4 expression and stability. (A) The expression of ferroptosis-related genes from RNA-sequencing. (B) The expression of ferroptosis-related genes in M16-knockdown or M16-overexpressing KMBC cells was confirmed by qPCR. (C) WB showed that the protein of ATF4 in M16-knockdown or M16-overexpressing KMBC and RBE cells. (D) The location of M16 in KMBC and EBE cells was shown by CLSM. (E-F) The m6A methylation level of ATF4 in KMBC cells transfected with si M16 (E), M16 plasmids (F) and controls. (G) Relative luciferase activity of ATF4-Wt and ATF4-Mut in M16-knockdown KMBC cells. (H) ATF4 mRNA levels in M16-knockdown and M16-overexpressing KMBC cells treated with actinomycin D. The data are presented as the mean ± SD. * p < 0.05, ** p < 0.01, *** p < 0.005.
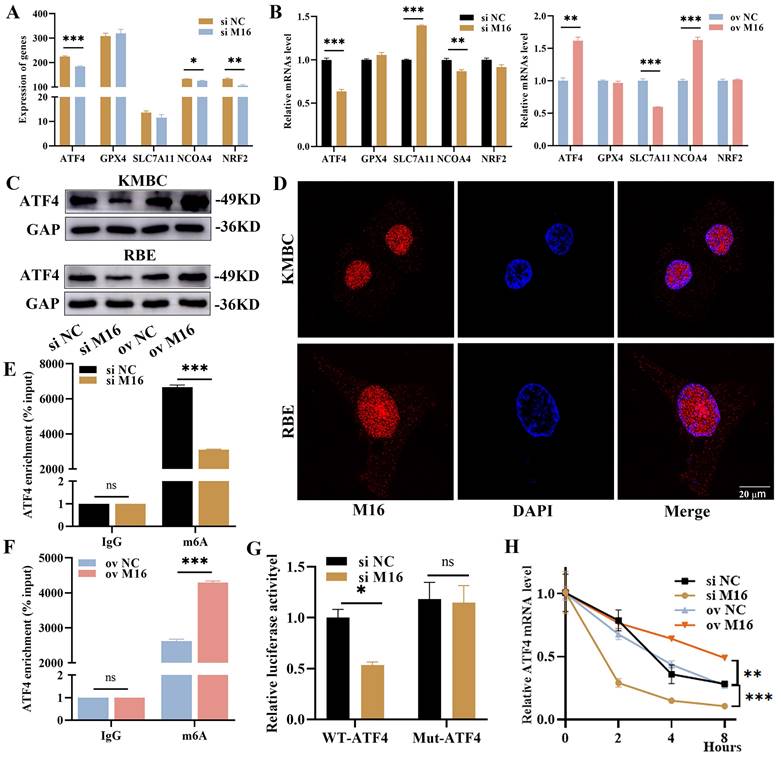
The way that METTL16 exerts functions hinges on its location in cells. In the nucleus, METTL16 works in m6A-dependent manner, while in the cytoplasm, it does in an m6A-independent manner [11, 31]. In CC tissues, KMBC and RBE cells, METTL16 was mainly localized in the nucleus (Figure 5D), suggesting that METTL16 takes effect primarily through m6A modification. Therefore, we postulated that ATF4 undergoes METTL16-mediated m6A modification and performed SRAMP analysis. The results manifested that there are three m6A modification sites on ATF4 mRNA, among which the second appeared to be the most likely (Figure S3D). This suggested that METTL16 may interpose the m6A modification of ATF4 mRNA, as evidenced by MeRIP (Figure S3E). Notably, METTL16 knockdown reduced the m6A level of ATF4, while METTL16 overexpression increased the m6A level of ATF4 (Figure 5E and F). Subsequently, dual-luciferase reporters were performed to further confirm the m6A site on ATF4 (Figure S3F). The results revealed that METTL16 knockdown significantly reduced the firefly luciferase in wild-type ATF4. However, METTL16 knockdown had no impact on the renilla luciferase activity from wild-type to mutant ATF4 (Figure 5G). Thus, the m6A modification of ATF4 is under the control of METTL16. It is widely acknowledged that m6A modification primarily regulates mRNA stability, leading to changes of mRNA expression [32]. We performed RNA stability experiments and found that METTL16 knockdown decreased ATF4 stability, whereas METTL16 overexpression increased ATF4 stability (Figure 5H, Figure S3G). Overall, these results suggest that METTL16 promotes expression and stability of ATF4 through m6A modification.
ATF4 promotes CC progress and rescues the effect of METTL16 in CC
Then, the role of ATF4 in CC was further explored. GEO analysis showed that ATF4 was upregulated in CC tissues (2/3 datasets), which was in line with HPA, indicating that ATF4 may play an oncogenic role in CC (Figure 6A, Figure S4A). In order to testify this notion, ATF4 knockdown and overexpression were carried out and verified in KMBC and RBE cells (Figure S4B and C). The EdU assays showed that ATF4 knockdown reduced proliferation in KMBC and RBE cells, whereas ATF4 overexpression increased proliferation (Figure S4D). These findings were in line with the results of colony formation (Figure 6B, Figure S4E). Furthermore, The Transwell assays manifested ATF4 knockdown reduced the migration of KMBC and RBE cells, while ATF4 overexpression increased migration (Figure S4F). Similarly, the migrative functions of ATF4 were further confirmed by wound-healing assays (Figure 6C, Figure S4G). To further verify the functions of ATF4 in METTL16-induced effects in CC, rescue experiments were performed. In EdU assays, ATF4 overexpression restored the reduced proliferative capacity caused by METTL16 knockdown in KMBC and RBE cells, whereas ATF4 knockdown abolished the increased proliferative capacity resulting from METTL16 overexpression (Figure 6D, Figure S5A). Similar migration results were obtained in Transwell assays (Figure 6E, Figure S5B). Together, these findings show that ATF4 facilitate the growth and revises METTL16-induced progression in CC.
ATF4 promotes the growth and rescues the effects of M16 in CC. (A) The expression of ATF4 in GSE107943, GSE76297 and GSE132305. (B-C) The proliferation and migration of ATF4-knockdown or ATF4-overexpressing KMBC cells were investigated by colony formation assays (B) and wound healing assays (C). (D-E) ATF4 knockdown/overexpression rescued the M16-overexpressing/ knockdown effect on proliferation (D) and migration (E) in KMBC cells. (F-G) ATF4 knockdown/overexpression reversed the M16-overexpressing/ knockdown effect on GSH (F) and MDA (G) levels in KMBC cells. The data are presented as the mean ± SD. * p < 0.05, ** p < 0.01, *** p < 0.005.
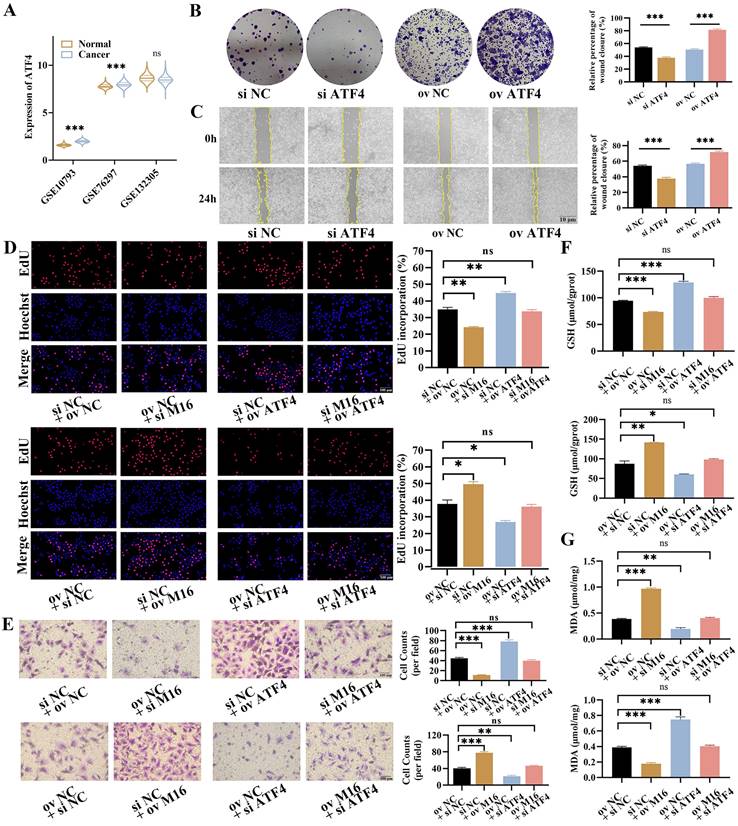
As known, ATF4 is a key regulatory gene in ferroptosis, which could inhibit ferroptosis and enhances tumor growth [22, 33]. Here, we found that ATF4 knockdown decreased intracellular GSH levels but elevated intracellular MDA levels, while ATF4 overexpression increased intracellular GSH levels but decreased intracellular MDA levels in KMBC and RBE cells (Figure S5C and D). In rescue assays, ATF4 overexpression reversed the METTL16 knockdown-induced changes in intracellular GSH and MDA levels in KMBC and RBE cells, while ATF4 knockdown restored the METTL16 overexpression-induced changes in intracellular GSH and MDA levels (Figure 6F and G, Figure S5E and F). Collectively, these observations demonstrate that ATF4 is responsible for METTL16-mediated regulation of ferroptosis in CC.
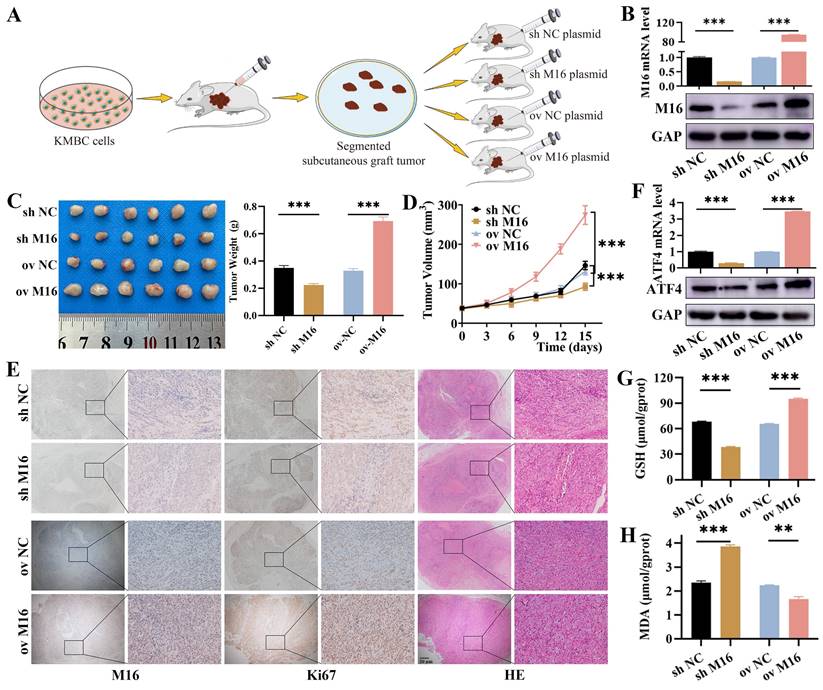
METTL16 inhibits ferroptosis in CC by ATF4 in mice
Based on the results from in vitro studies, nude mouse xenograft models (Figure 7A) with METTL16 knockdown and overexpression plasmids were constructed to validate the role of METTL16 in CC in vivo, and the effectiveness of METTL16 overexpression and knockdown was confirmed by qPCR, WB and IHC (Figure 7B and E). As shown, METTL16 knockdown reduced tumor growth and weight, whereas METTL16 overexpression increased tumor growth and weight (Figure 7C and D). Furthermore, we used Ki-67 and HE staining to assess whether cell proliferation and necrosis were affected by METTL16 knockdown and overexpression. Our results revealed that METTL16 knockdown reduced proliferation but increased necrosis, while METTL16 overexpression enhanced proliferation but suppressed necrosis (Figure 7E). Regarding to ferroptosis, we found METTL16 knockdown decreased the expression of ATF4, while METTL16 overexpression increased the expression of ATF4 (Figure 7F). Then, we tested indicator of ferroptosis in tissues and found that METTL16 knockdown decreased GSH levels but increased MDA. Conversely, METTL16 overexpression elevated GSH levels but decreased MDA (Figure 7G and H). Taken together, these results indicate that METTL16 promotes progression and ferroptosis in CC in vivo.
M16 inhibits ferroptosis in CC via ATF4 in vivo. (A) M16 overexpression or knockdown xenograft model in nude mice. (B) Xenograft tumors with M16 knockdown or overexpression were confirmed by qPCR and WB. (C-D) The tumor weight (C) and volume (D) of the xenografts were measured. (E) Xenograft tumors were stained for M16, Ki-67, and HE. (F) qPCR and WB showed that the expression of ATF4 in the xenograft tumors with M16 knockdown or overexpression. (G-H) GSH (G) and MDA (H) levels in the xenograft tumors were measured. The data are presented as the mean ± SD. ** p < 0.01, *** p < 0.005.
Discussion
In recent years, there have been great advancements in targeted drugs and immunotherapy, but the prognosis of patients with CC remains poor. Thus, more attention should be paid to new therapeutics. In this study, we found the potential of METTL16 as a treatment for CC and revealed a novel mechanism for improving anti-tumor effects by inducing ferroptosis. Specifically, METTL16 is upregulated and enhances ATF4 mRNA stability and expression via m6A modification, further counteracting ferroptosis and promoting the growth of CC.
Among the approximate 150 post-transcriptional RNA modifications that have been identified, m6A modifications are the most prevalent [34, 35]. m6A modifications play oncogenic or tumor-suppressive roles in cancer by regulating RNA splicing, translocation, degradation, stability, and translation. As a new m6A methyltransferase, METTL16 not only regulates MAT2A expression in response to yield of s-adenosyl methionine (SAM) that is an essential cofactor for methylation, but also binds to MAT2A to form a METTL16-RNA complex, which consequently drives downstream RNA methylation [36-38]. Here, we found that METTL16 is highly expressed in CC and correlated with poor prognosis. Whatmore, METTL16 overexpression enhanced the migration and proliferation of KMBC and RBE cells, while METTL16 knockdown inhibited these biological behaviors, similar to the results in vivo. These data manifested that METTL16 has tumor-promoting impacts on CC, which is consistent with previous reports in hepatocellular carcinoma, colorectal carcinoma, gastric cancer and lung cancer [38-41]. In contrast, METTL16 suppresses cell proliferation through the p21 pathway in pancreatic adenocarcinoma [42]. These contradictory functions in different cancers may have occurred due to tissue specificity and cancer specificity.
Metabolic reprogramming is a central hallmark of cancers, and METTL16 has a great impact on metabolic reprogramming, such as branched chain amino acid metabolism and glucose metabolism [41, 43]. We also found that METTL16 is mainly associated with oxidative stress, as confirmed by measuring ROS levels. Ferroptosis is activated by oxidative stress that results from glutathione depletion, hemin, and/or reactive lipid species [44, 45]. Thereafter, we explored whether METTL16 affects the process of ferroptosis. Morphologically, METTL16 knockdown caused mitochondria to become smaller with cristae disappearing, and decreased the MMP, indicating that METTL16 acts against ferroptosis in CC. Mechanistically, METTL16 knockdown decreased intracellular GSH levels and increased MDA, Fe2+ and LPO levels, similar to the findings in breast cancer [46]. METTL16 overexpression elevated intracellular GSH levels and reduced MDA, Fe2+ and LPO levels. These results suggest that METTL16 facilitates the progression of CC by suppressing ferroptosis.
In response to the mechanism of METTL16 in regulating ferroptosis, previous studies have shown three different ways to improve METTL16-mediated resistance to ferroptosis: (i) SAM, ascended by METTL16, ultimately gives rise to cysteine via the trans-sulfuration pathway, which recures lipid peroxidation and impedes [47,48]; (ii) SAM also increases H3K4me3 modification on the promoter of ACSL3 and subsequently upregulates its expression, protecting against ferroptosis [49]; and (iii) METTL16 promotes the expression and stability of GPX4 to further confront ferroptosis [46]. Here, another novel way from our research was that METTL16 relies on ATF4 to inhibit ferroptosis in an m6A-dependent manner. First, we found that METTL16 is primarily localized in the nucleus and to a lesser extent in the cytoplasm. In the nucleus, METTL16 is involved in the m6A modification of target RNAs, while in the cytoplasm, METTL16 masters cell fate through translation in an m6A-independent manner [11, 31]. These data mirrored that METTL16 exerts its effect mainly via m6A modification. Next, according to RNA-sequencing, ATF4 was confirmed to be another downstream target of METTL16 and to be modified by METTL16-mediated m6A methylation, as confirmed by MeRIP and dual-luciferase reporter assays. Furthermore, RNA stability assays showed that the stability of ATF4 mRNA is significantly reduced after METTL16 knockdown, but that increased after METTL16 overexpression. Overall, these data provide strong evidence that METTL16 promotes the expression and stability of ATF4 to curb ferroptosis via m6A modification.
ATF4 is an important regulator in response to hypoxia or amino acid restriction [50, 51]. Most of its target genes are involved in various salvage pathways that promote cell survival, but others prepare the cell for death [52]. In hepatocellular carcinoma, renal cell carcinoma and pancreatic ductal adenocarcinoma, ATF4 suppresses ferroptosis and promotes cell proliferation through the ATF4/SLC7A11 and ATF4/HSPA5/GPX4 axes [22, 23, 33]. Conversely, ATF4 facilitates apoptosis and exerts anti-tumor effects via the ATF4/CHOP axis in the treatment of multiple myeloma with aspirin [53]. As to CC, the function of ATF4 is not yet fully understood. Our study first showed that ATF4 is highly expressed in CC and accelerates migration and proliferation by inhibiting ferroptosis. Moreover, ATF4 knockdown abolished the effects of METTL16 overexpression on proliferation and ferroptosis, and ATF4 overexpression reversed the effects of METTL16 knockdown. Based on these findings, it is confirmed that METTL16/ATF4 performs important functions on ferroptosis in CC. As known, ATF4 is a common upstream regulator of both SLC7A11 and GPX4 those inhibit ferroptosis by producing GSH and detoxifying lipid peroxidation, respectively [21-23]. However, whether both SLC7A11 and GPX4 are affected by METTL16/ATF4 remains unknown, and further experiments are needed.
Our work elucidates the role of METTL16 in CC and its ability to inhibit ferroptosis. METTL16 is upregulated in CC tissues, conferring a poor prognosis for patients. Mechanistically, METTL16 promotes the expression and stability of ATF4, thereby suppressing ferroptosis in CC and ultimately accelerating tumor growth. These findings provide valuable insights into METTL16-mediated m6A modifications in CC as well as effective therapeutic strategies for CC.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We would like to thank the Academy of Medical Sciences of Zhengzhou University Translational Medicine platform and the Department of Zhengzhou University Animal Center for providing the experimental equipment.
Funding
This work was supported by National Natural Science Foundation of China (81870457 and 82172944); Key science and technology projects of Henan Province (232102311048).
Ethics approval and consent to participate
All protocols using human specimens were approved by the Ethics Committee of The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China and performed in compliance with the principles of the Declaration of Helsinki. All participants in the study provided informed consent. All animal experiments were approved by Animal Ethics Committee of Zhengzhou University.
Availability of data and material
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
Senfeng Zhao, Jiahui Cao and Ruopeng Liang performed all the experimental assays and wrote the original draft. Tingting Peng, Shitao Wu and Zhipu Liu performed bioinformatics analyses. Senfeng Zhao, Yahui Wu, Liming Song and Chenguang Sun performed data analysis and statistics. Yin Liu, Junmou Gu and Libo Wang contributed to manuscript revisiting. Rongtao Zhu, Weijie Wang and Yuling Sun provided constructive comments and discussion. Yuling Sun conceptualized and supervised the study.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bertuccio P, Malvezzi M, Carioli G. et al. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma. J Hepatol. 2019;71:104-14
2. Banales JM, Marin J, Lamarca A. et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol. 2020;17:557-88
3. Izquierdo-Sanchez L, Lamarca A, La Casta A. et al. Cholangiocarcinoma landscape in Europe: Diagnostic, prognostic and therapeutic insights from the ENSCCA Registry. J Hepatol. 2022;76:1109-21
4. Merters J, Lamarca A. Integrating cytotoxic, targeted and immune therapies for cholangiocarcinoma. J Hepatol. 2023;78:652-57
5. Yang F, Hilakivi-Clarke L, Shaha A. et al. Metabolic reprogramming and its clinical implication for liver cancer. Hepatology. 2023;78:1602-24
6. Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71:3971-75
7. Wang X, Lu Z, Gomez A. et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117-20
8. Deng X, Su R, Weng H. et al. RNA N6-methyladenosine modification in cancers: current status and perspectives. Cell Res. 2018;28:507-17
9. Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20:608-24
10. Shang A, Gu C, Wang W. et al. Exosomal circPACRGL promotes progression of colorectal cancer via the miR-142-3p/miR-506-3p- TGF-β1 axis. Mol Cancer. 2020;19:117
11. Qi YN, Liu Z, Hong LL. et al. Methyltransferase-like proteins in cancer biology and potential therapeutic targeting. J Hematol Oncol. 2023;16:89
12. Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12:31-46
13. Stockwell BR, Friedmann Angeli JP, Bayir H. et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171:273-85
14. Xie Y, Hou W, Song X. et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369-79
15. Zhou H, Zhou YL, Mao JA. et al. NCOA4-mediated ferritinophagy is involved in ionizing radiation-induced ferroptosis of intestinal epithelial cells. Redox Biol. 2022;55:102413
16. Tang D, Chen X, Kang R. et al. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107-25
17. Zhang C, Liu X, Jin S. et al. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol Cancer. 2022;21:47
18. Qu X, Liu B, Wang L. et al. Loss of cancer-associated fibroblast-derived exosomal DACT3-AS1 promotes malignant transformation and ferroptosis-mediated oxaliplatin resistance in gastric cancer. Drug Resist Updat. 2023;68:100936
19. Sun X, Ou Z, Chen R. et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173-84
20. Mao C, Liu X, Zhang Y. et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586-90
21. Lewerenz J, Sato H, Albrecht P. et al. Mutation of ATF4 mediates resistance of neuronal cell lines against oxidative stress by inducing xCT expression. Cell Death Differ. 2012;19:847-58
22. He F, Zhang P, Liu J. et al. ATF4 suppresses hepatocarcinogenesis by inducing SLC7A11 (xCT) to block stress-related ferroptosis. J Hepatol. 2023;79:362-77
23. Zhu S, Zhang Q, Sun X. et al. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017;77:2064-77
24. Ziegler D, Sohr CG, Nourooz-Zadeh J. Oxidative stress and antioxidant defense in relation to the severity of diabetic polyneuropathy and cardiovascular autonomic neuropathy. Diabetes Care. 2004;27:2178-83
25. Wang W, Duan J, Ma W. et al. Trimanganese Tetroxide Nanozyme protects Cartilage against Degeneration by Reducing Oxidative Stress in Osteoarthritis. Adv Sci (Weinh). 2023;10:e2205859
26. Wu CY, Cilic A, Pak O. et al. CEACAM6 as a Novel Therapeutic Target to Boost HO-1-mediated Antioxidant Defense in COPD. Am J Respir Crit Care Med. 2023;207:1576-90
27. Zhu Y, Wang K, Jia X. et al. Antioxidant peptides, the guardian of life from oxidative stress. Med Res Rev. 2024;44:275-364
28. Shen Q, Liang M, Yang F. et al. Ferroptosis contributes to developmental cell death in rice blast. New Phytol. 2020;227:1831-46
29. Zhang Z, Guo M, Li Y. et al. RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy. 2020;16:1482-505
30. Jiang Y, Zhao J, Li R. et al. CircLRFN5 inhibits the progression of glioblastoma via PRRX2/GCH1 mediated ferroptosis. J Exp Clin Cancer Res. 2022;41:307
31. Su R, Dong L, Li Y. et al. METTL16 exerts an m6A-independent function to facilitate translation and tumorigenesis. Nat Cell Biol. 2022;24:205-16
32. Musselman CA, Lalonde ME, Côté J. et al. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol. 2012;19:1218-27
33. Kang L, Wang D, Shen T. et al. PDIA4 confers resistance to ferroptosis via induction of ATF4/SLC7A11 in renal cell carcinoma. Cell Death Dis. 2023;14:193
34. Dominissini D, Moshitch-Moshkovitz S, Schwartz S. et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201-06
35. Sendinc E, Shi Y. RNA m6A methylation across the transcriptome. Mol Cell. 2023;83:428-41
36. Pendleton KE, Chen B, Liu K. et al. The U6 snRNA m6A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell. 2017;169:824-35.e14
37. Doxtader KA, Wang P, Scarborough AM. et al. Structural Basis for Regulation of METTL16, an S-Adenosylmethionine Homeostasis Factor. Mol Cell. 2018;71:1001-11.e4
38. Dai YZ, Liu YD, Li J. et al. METTL16 promotes hepatocellular carcinoma progression through downregulating RAB11B-AS1 in an m6A-dependent manner. Cell Mol Biol Lett. 2022;27:41
39. Wang XK, Zhang YW, Wang CM. et al. METTL16 promotes cell proliferation by up-regulating cyclin D1 expression in gastric cancer. J Cell Mol Med. 2021;25:6602-17
40. Wang F, Zhang J, Lin X. et al. METTL16 promotes translation and lung tumorigenesis by sequestering cytoplasmic eIF4E2. Cell Rep. 2023;42:112150
41. Wei W, Zhang ZY, Shi B. et al. METTL16 promotes glycolytic metabolism reprogramming and colorectal cancer progression. J Exp Clin Cancer Res. 2023;42:151
42. Xie F, Zheng Y, Fu W. et al. The m6A methyltransferase METTL16 inhibits the proliferation of pancreatic adenocarcinoma cancer cells via the p21 signaling pathway. Front Oncol. 2023;13:1138238
43. Han L, Dong L, Leung K. et al. METTL16 drives leukemogenesis and leukemia stem cell self-renewal by reprogramming BCAA metabolism. Cell Stem Cell. 2023;30:52-68.e13
44. Ratan RR. The Chemical Biology of Ferroptosis in the Central Nervous System. Cell Chem Biol. 2020;27:479-98
45. Panda SK, Peng V, Sudan R. et al. Repression of the aryl-hydrocarbon receptor prevents oxidative stress and ferroptosis of intestinal intraepithelial lymphocytes. Immunity. 2023;56:797-812.e4
46. Ye F, Wu J, Zhang F. METTL16 epigenetically enhances GPX4 expression via m6A modification to promote breast cancer progression by inhibiting ferroptosis. Biochem Biophys Res Commun. 2023;638:1-6
47. Morellato AE, Umansky C, Pontel LB. The toxic side of one-carbon metabolism and epigenetics. Redox Biol. 2021;40:101850
48. Homma T, Kobayashi S, Fujii J. Methionine Deprivation Reveals the Pivotal Roles of Cell Cycle Progression in Ferroptosis That Is Induced by Cysteine Starvation. Cells. 2022;11:1603
49. Ma M, Kong P, Huang Y. et al. Activation of MAT2A-ACSL3 pathway protects cells from ferroptosis in gastric cancer. Free Radic Biol Med. 2022;181:288-99
50. Pathria G, Verma S, Yin J. et al. MAPK signaling regulates c-MYC for melanoma cell adaptation to asparagine restriction. EMBO Rep. 2021;22:e51436
51. Verginadis II, Avgousti H, Monslow J. et al. A stromal Integrated Stress Response activates perivascular cancer-associated fibroblasts to drive angiogenesis and tumour progression. Nat Cell Biol. 2022;24:940-53
52. Wortel I, van der Meer LT, Kilberg MS. et al. Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol Metab. 2017;28:794-806
53. Liu H, Xiong C, Liu J. et al. Aspirin exerts anti-tumor effect through inhibiting Blimp1 and activating ATF4/CHOP pathway in multiple myeloma. Biomed Pharmacother. 2020;125:110005
Author contact
![]() Corresponding author: Yuling Sun, Department of Hepatobiliary and Pancreatic Surgery, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, 450052, China. Email: ylsunedu.cn.
Corresponding author: Yuling Sun, Department of Hepatobiliary and Pancreatic Surgery, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, 450052, China. Email: ylsunedu.cn.