Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Dual CAR structures
Dual-targeted CAR-T cell therapy...
Discussion and Prospects
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(6):2676-2691. doi:10.7150/ijbs.108036 This issue Cite
Review
From molecular design to clinical translation: dual-targeted CAR-T strategies in cancer immunotherapy
Zhenrong Wang1*, Mengyi Wang1*, Mengting Wang1, Ruijie Zhou1, Xiaotong Deng1, Xin Ouyang1, Minghui Chu1, Xinyu Wei1, Lei Yang4, Jinbiao Liu2,3 ![]() , Yao Xu1
, Yao Xu1 ![]()
1. Institute of Biology and Medicine, College of Life Science and Health, Wuhan University of Science and Technology, Wuhan, Hubei 430081, China.
2. National “111” Center for Cellular Regulation and Molecular Pharmaceutics, Key Laboratory of Fermentation Engineering (Ministry of Education), Hubei Provincial Cooperative Innovation Center of Industrial Fermentation, Hubei Key Laboratory of Industrial Microbiology, Sino-German Biomedical Center, Hubei University of Technology, Wuhan, Hubei, 430068, China.
3. Institute of Medical Microbiology, Jinan University, Guangzhou 510632, China.
4. People's Hospital of Jingyang County, Xianyang, Shaanxi, 713700, China.
* These authors contributed equally to this work.
Received 2024-12-2; Accepted 2025-3-10; Published 2025-3-31
Abstract

The pathogenesis of tumors involves various abnormalities at both the cellular and genetic levels. Chimeric antigen receptor (CAR)-T cell immunotherapy has emerged as a transformative treatment strategy that effectively addresses these challenges. While CAR-T therapy has shown remarkable success in treating hematological malignancies, limitations have been identified, particularly in single antigen-targeting CAR-T therapies. These limitations include antigenic mutation or loss, reduced efficacy against leukemia, and poor results in solid tumors due to factors like low CAR-T cell persistence, limited tumor infiltration, rapid cell exhaustion, the suppressive tumor microenvironment, and heterogeneous tumor antigen expression. In recent years, multi-antigen targeted CAR-T therapies have garnered significant attention for their potential to prevent tumor relapse and progression. This review outlines the fundamental design of dual CAR structures and summarizes the major advancements in both preclinical studies and clinical trials of dual-targeted CAR-T cell therapy, categorized by cancer type. Additionally, it discusses the challenges associated with dual-targeted CAR-T therapy and the strategies to enhance its efficacy and applicability in treating both hematologic and solid tumors. In conclusion, the progress in dual-targeted CAR-T cell therapy presents a promising therapeutic avenue for multiple malignancies, offering insights into future modifications of immunotherapy to advance the field.
Keywords: dual target, CAR-T cell therapy, leukemia, solid tumor, immunotherapy
Introduction
Cancer remains one of the most pressing global health challenges, with an estimated 20 million new cases and 9.7 million deaths reported in 2022 [1], primarily due to its high metastasis rate and poor prognosis. Despite significant advancements in medical technology and clinical strategies, including surgery, radiotherapy, and chemotherapy, several limitations still impact therapeutic outcomes. These limitations include severe side effects, high recurrence rates, and the development of drug resistance [2, 3]. In recent years, cell immunotherapy, which harnesses and enhances the immune system to target and eliminate cancer cells, has emerged as a promising cancer treatment approach, demonstrating immense potential and unique advantages for reducing relapse rate [4].
Chimeric antigen receptor T (CAR-T) cell therapy has revolutionized cancer treatment, particularly for hematological malignancies. With over 80% complete remission rates in relapsed/refractory B-cell acute lymphoblastic leukemia (B-ALL) and 40-50% long-term survival rates in diffuse large B-cell lymphoma (DLBCL), CAR-T therapy has demonstrated unprecedented success where conventional therapies have failed [5, 6]. This groundbreaking approach leverages genetically engineered T cells to precisely target and eliminate cancer cells, offering a paradigm shift in immunotherapy. However, the clinical application of single-antigen targeted CAR-T cells faces significant challenges, including antigen escape, therapy-related toxicities (e.g., cytokine release syndrome), and limited efficacy in solid tumors [7, 8].
Given these limitations, recent evidence has proposed dual-CAR-T cell therapies, which simultaneously express two distinct CAR structures on a single T cell, enabling the modified CAR-T cells to recognize two different antigens on tumor cells [9, 10]. Compared to single-target CAR-T therapy, the dual-targeting strategy can enhance the specificity and effectiveness of cancer therapy by reducing the risk of antigen escape and promoting immune cell infiltration [11]. This article reviews the structural properties of dual-targeted CAR-T cells and highlights the potential advantages of dual CAR over single CAR structure. Moreover, we discuss recent innovations in preclinical studies and clinical trials for both hematological malignancies and solid tumors, as well as the challenges and future prospects for enhancing the efficacy and applicability of dual-targeted CAR-T cell therapy in treating cancer.
This review aims to provide a comprehensive overview of dual-targeted CAR-T cell therapies. The review begins by describing three main types of dual-targeted CAR-T structures. We then summarize the research progress of dual-targeted CAR-T therapies in hematologic malignancies and solid tumors, respectively. Finally, we address the challenges associated with the application of dual-targeted CAR-T cells and propose potential strategies, while also discussing the future prospects for their broader clinical translation.
Overview of CAR-T therapeutic issues
CAR-T cell therapy has shown significant therapeutic effects in treating B-cell malignancies. However, in some patients, tumor cells can evade CAR-T cell attacks through multiple mechanisms, including antigenic modulation, epitope masking, and lineage switching. For instance, in pediatric B-cell acute lymphoblastic leukemia (B-ALL), CD19-negative relapse occurs in approximately 10-30% of patients post-CAR-T therapy due to CD19 downregulation or loss, often mediated by alternative splicing of CD19 mRNA or clonal selection of pre-existing CD19-negative subpopulations [12]. The mutation or loss of tumor antigens is a prevalent mechanism of resistance to single-target CAR-T cell therapy in B-cell malignancies. In solid tumors, the limitations of single-target CAR-T therapy are even more pronounced due to the high heterogeneity of tumor antigens and the complex tumor microenvironments (TME), which is composed of a range of immunosuppressive factors, including regulatory T cells, myeloid-derived suppressor cells, and tumor-associated macrophages, all of which can dampen CAR-T cell function [13]. Overall, the challenges of single-target CAR-T cell therapy are multifaceted and can be attributed to various factors, including the antigen escape and tumor heterogeneity, immunosuppressive TME, limited CAR-T cell persistence and exhaustion, and therapy-related toxicities [8, 14-17]. These limitations underscore the need for multi-antigen targeting strategies, such as dual-targeted CAR-T cells, to overcome antigen escape and enhance therapeutic efficacy.
Advantages of dual-targeted CAR-T therapy relative to single CAR
Dual-targeted CAR-T cell therapy is an innovative approach in cancer treatment that involves engineering CAR-T cells to express two distinct antigen recognition domains, enabling them to target two different antigens on tumor cells simultaneously. This therapeutic strategy offers several advantages over traditional single-target CAR-T cell therapy [9, 18-21]. (1) Reduced potential for antigen escape: By targeting two antigens concurrently, dual-targeted CAR-T cell therapy reduces the likelihood of tumor cells evading immune attack through the loss or downregulation of a single antigen. (2) Enhanced therapeutic efficacy: Dual-targeted CAR-T cells can recognize and attack tumor cells via two distinct mechanisms, thereby amplifying the overall antitumor activity and potentially leading to more effective treatment outcomes. (3) Improved treatment specificity: The specificity of clinical therapy is significantly increased by targeting two antigens, which helps minimize damage to normal cells and may reduce the side effects commonly associated with traditional cancer treatments. (4) Potential synergistic effects: The combination of two CAR structures can generate synergistic interactions, enhancing cytotoxicity and improving the efficiency of tumor cell elimination. (5) Broader application range: Dual-targeted CAR-T cell therapy may be applicable to a wider range of tumor types, particularly those with insufficient single antigen expression or exhibiting antigen heterogeneity.
Dual CAR structures
Dual-targeted CAR-T cell therapy, which introduces two distinct chimeric antigen receptors (CARs) on a single T cell, has emerged as an innovative strategy for cancer treatment. These bispecific CAR constructs are designed to enable T cells to simultaneously recognize and target two different antigens on tumor cells, thereby enhancing both the specificity and potency of the immune response. The structural design of dual CAR-T cells can be primarily categorized into three types: tandem, parallel, and synNotch configurations. Tandem CAR-T cells feature a single expression unit containing two CAR domains in tandem, allowing T cells to activate a cascade of amplification signals in response to the second antigen after the initial antigen recognition of the first antigen (Figure 1A). In contrast, parallel CAR-T cells express two independent CAR structures on the same T cell, permitting simultaneous recognition and targeting of two distinct antigens (Figure 1B). The synNotch structure mimics the signaling mechanism of the natural T cell receptor, enabling selective signal activation in dual CAR-T cells upon recognition of a specific antigen (Figure 1C). These diverse structural designs reflect the ongoing exploration and innovation by researchers to enhance the safety of dual CAR-T cell therapy, reduce toxicity, and improve therapeutic efficacy. The differences among the three CAR structures, such as target recognition, signaling pathways, and potential advantages, were summarized in Table S1. In the following sections, we will thoroughly explore the characteristics, advantages, and clinical potential of the three configurations.
The schematic structures of dual-targeted CAR-T cells. (A) Tandem dual CAR structure. (B) Parallel dual CAR structure. (C) synNotch dual CAR structure.
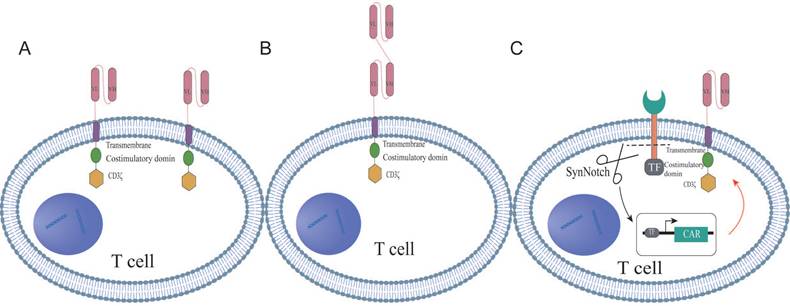
Dual CAR tandem structures
The second-generation CAR structure is modified to construct the tandem dual CAR molecule, which consists of two distinct antigen recognition domains targeting different epitopes, along with a spacer sequence, a transmembrane domain, a co-stimulatory domain, and a signaling domain. Specifically, the antigen recognition domains are typically single-chain variable fragments (scFvs); the spacer sequence connects the dual scFvs to the co-stimulatory and signaling domains, often utilizing the hinge region of CD8α; the transmembrane domains are primarily derived from CD8α and CD28; while the co-stimulatory domains commonly include CD28 and 4-1BB; CD3ζ serves as the signaling domain. Most dual-targeting scFvs are linked by a unique connector, with the glycine-serine linker (Gly4Ser) being the most widely used [22, 23]. This linker, composed of repetitive glycine and serine residues, provides a flexible bridge that reduces steric hindrance, allowing the connected proteins to fold and function independently. This flexibility enables the heavy and light chains, linked by the Gly4Ser linker, to form a complete antibody structure, optimizing antigen recognition and binding. Previous studies have also incorporated a reporter gene after CD3ζ through a T2A cleavage site to assess the transduction efficiency of the engineered cells. For example, Schmidts et al. [20] linked a mCherry reporter gene, while Dai et al. [22] connected EGFRt to the CAR fragment via a T2A sequence.
Dual CAR parallel structures
The parallel dual CAR molecular structure utilizes a second-generation design, featuring two distinct receptor targets that are independently expressed on T cells. This structure sequentially comprises the antigen recognition domains for two different targets, a hinge region, a transmembrane domain, co-stimulatory domains, and a signaling domain [24, 25]. The antigen recognition domains are primarily composed of scFvs, while the hinge region connects the scFvs of the two distinct targets to the co-stimulatory and signaling domains, typically utilizing the hinge region of CD8α to optimize spatial flexibility and antigen-binding efficiency [26]. The transmembrane domains are predominantly derived from CD8α and CD8, with CD28 also serving as a transmembrane domain. The common co-stimulatory domains include CD28 and 4-1BB4, and CD3ζ functions as the signaling domain. In parallel CAR design, CD28 and 4-1BB are frequently used as co-stimulatory domains to activate T cells, playing a pivotal role in T cell activation and proliferation. Each co-stimulatory unit is independently expressed and strategically positioned near the T cell plasma membrane, thereby mimicking the natural arrangement of T cell receptors (TCRs) and co-stimulatory receptors under physiological conditions [27]. Previous studies have reported that other co-stimulatory domains, such as CD137 and ICOS, can enhance CAR-T cell persistence and effector functions by coordinately activating metabolic pathways [28, 29]. The hinge region can consist of either the CD8 hinge or the IgG4mt hinge, while the signaling domain is most commonly CD3ζ. In research involving parallel dual-targeted CAR-T cells, investigators have adjusted the order of scFvs and the signaling domains to enable a single T cell to simultaneously recognize and target two distinct tumor-associated antigens [30].
Dual CAR synNotch structures
In the synNotch structure, the antigen-specific scFv is linked to the Notch core and transcription factor. In general, synNotch receptors contain an N-terminal CD8α signal peptide (MALPVTALLLPLALLLHAARP) for membrane targeting, as well as an α-myc tag (EQKLISEEDL) or flag tag (DYKDDDDK) or GFP tag or BFP tag for detecting surface expression [31]. However, some synNotch receptors are engineered by attaching the humanized antigen scFv to the intracellular Notch core domain, which is fused with the tTA transcription factor [32]. The intracellular domain of the synNotch structure may also include a fusion protein comprising a DNA-binding domain and transcription activator, with the upstream activation sequence (UAS) integrated into the receptor's structural elements. Additionally, markers such as blue fluorescent protein (BFP) are incorporated into T cells for efficient cell sorting [33].
In conclusion, the tandem structure of dual-targeted CAR-T cells positions two CAR domains in series within a single expression unit, allowing T cells to first recognize the initial antigen and then, through a cascade amplification mechanism, activate a response to the second antigen. This design enables T cells to simultaneously recognize and target two distinct tumor antigens, thereby enhancing their ability to identify and eliminate tumor cells [34]. In contrast, the parallel structure of CAR-T cells expresses two independent CARs on the same T cell, enabling simultaneous recognition and attack of two different antigens. The design of parallel CAR-T cells is of significant importance, as it not only provides potent anti-tumor activity but also helps mitigate T cell exhaustion and senescence, thereby improving the longevity and functionality of T cells [35]. The synNotch CAR represents an innovative CAR structure that mimics the signaling mechanism of natural Notch receptors, allowing precise control over T cell activity. It combines two distinct antigen-binding domains, where recognition of the first antigen triggers a Notch receptor-like cleavage and release, thereby activating the expression of the second CAR and facilitating the recognition and response to the second antigen [36]. The synNotch CAR circuit, which targets highly specific solid tumor antigens, enhances both specificity and therapeutic efficacy by modulating T cell exhaustion. This approach not only improves specificity through multi-antigen sensing but also provides a universal strategy for enhancing efficacy through cell-autonomous and context-dependent regulation of CAR expression. A previous in vivo study revealed that the synNotch CAR significantly maintained T cell memory subset by preventing tonic signaling, which is crucial for the durability and sustained activity of cell therapies [37].
Dual-targeted CAR-T cell therapy in oncology
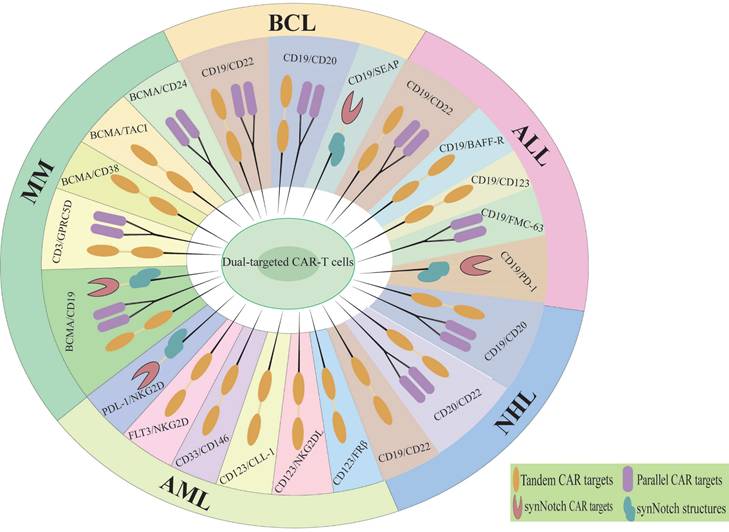
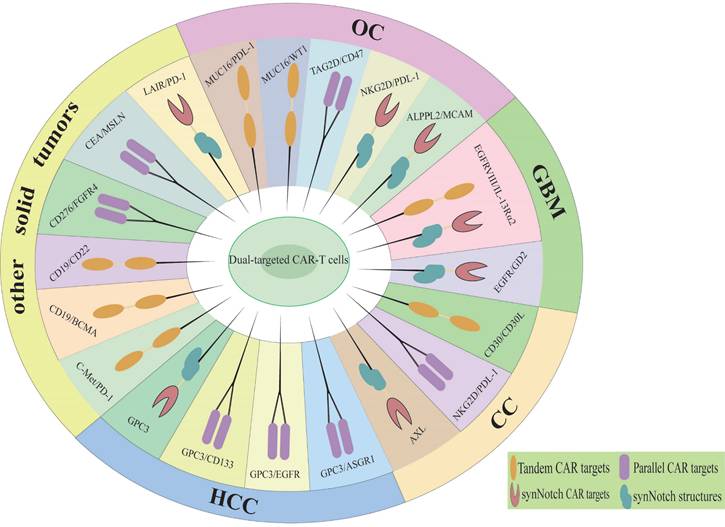
Dual-targeted CAR-T cells represent a promising strategy to address the limitations of single-target therapies, particularly in overcoming resistance mechanisms such as antigen escape, TME suppression, and T cell exhaustion [38]. By simultaneously targeting two tumor-associated antigens, dual-targeted CAR-T cells significantly reduce the risk of antigen loss or downregulation, a major cause of relapse in single-target therapies. In solid tumors, dual-targeting strategies that co-target tumor antigens and TME components have shown enhanced T cell infiltration and reduced immunosuppression [39]. Additionally, the incorporation of optimized co-stimulatory domains (e.g., CD28 and 4-1BB) improves T cell persistence and metabolic fitness, mitigating exhaustion [11]. The main diseases and dual targets for dual CAR-T cell therapy in hematological malignancies are illustrated in Figure 2 and Table 1. Although the application of dual CAR-T cell therapy remains in the early stages of exploration, studies have shown favorable safety profiles and preliminary antitumor activity across various solid tumor types (Figure 3, Table 2), including ovarian cancer and hepatocellular carcinoma. The results of these preclinical and clinical trials provide a strong foundation for expanding the use of dual CAR-T cell therapy to a broader range of cancers and open new avenues for future therapeutic strategies.
Dual therapeutic targets of CAR-T cell therapy in hematologic malignancies. MM, Multiple myeloma; BCL, B-cell lymphoma; ALL, Acute lymphoblastic leukemia; NHL, Non-hodgkin lymphoma; AML, Acute Myeloid Leukemia.
The outcomes of clinical trials of dual-targeted CAR-T therapy in hematological malignancies.
| Disease | dual-target | ClinicalTrials. number | Phase | Number of subjects | Complete remission rate (CR) | Overall survival rate (OS) | Ref. |
|---|---|---|---|---|---|---|---|
| B-NHL | CD19/CD22 | ChiCTR1800015575 | Phase I | 16 | 62.5% | 77.3% | [19] |
| B-NH | CD3/CD20 | NCT03075696 | Phase I | 177 | 36.8% | / | [40] |
| BCL | CD19/CD20 | NCT03233854 | Phase I | 21 | / | 77% | [41] |
| BCL | CD19/CD22 | NCT03289455 | Completed | 52 | 17% | / | [9] |
| BCL | CD19/CD22 | ChiCTR2100052247 | Completed | 24 | / | 90% | [42] |
| BCL | CD3/CD20 | NCT03625037 | Phase I/II | 157 | 38.9% | / | [43] |
| MM | BCMA/CD19 | ChiCTR2000033567 | Phase I/II | 50 | / | / | [44] |
| MM | BCMA/CD19 | NCT02546167 | Phase I | 30 | / | / | [45] |
| ALL | CD19/CD22 | NCT02443831 | Phase I | 12 | / | 75% | [46] |
| ALL | CD19/CD22 | NCT04227015 | Phase I | 6 | 83.3% | / | [47] |
| ALL | CD19/CD22 | NCT03289455 | Phase I | 15 | 86% | 60% | [48] |
| ALL | CD19/CD3 | NCT02013167 | Phase III | 271 | 12% | / | [49] |
Notes: B-NHL: B-cell Non-Hodgkin Lymphoma; BCL: B‑Cell Lymphoma; BCMA: B-Cell Maturation Antigen; MM: Multiple Myeloma; ALL:Acute Lymphoblastic Leukemia.
Dual therapeutic targets of CAR-T cell therapy in solid tumors. HCC, Hepatocellular carcinoma; CC, Cervical cancer; GBM, Glioblastoma multiforme; OC, Ovarian cancer.
The outcomes and efficacy of dual-targeted CAR-T therapy in tumor-bearing mouse models.
| Disease | Dual-target | Mouse model | Groups of mice | Single-CAR mouse survival rate | Double-CAR mouse survival rate | Tumor volume changes in single CAR mouse | Tumor volume changes in double CAR mouse | Ref. |
|---|---|---|---|---|---|---|---|---|
| HCC | GPC3/FAP | HepG2 CDX | 5 | 40%(day50) | 60%(day50) | Increase | Increase slowly | [39] |
| HCC | GPC3/PD-1 | TX | 4 | 80%(day40) | 100%(day40) | Increase | Decrease | [93] |
| HCC | GPC3/ CD133 | Huh-7 | 5 | 0(day75) | 80%(day75) | No Change | Decrease | [94] |
| HCC | GPC3/EGFR | Huh-7-luc | 5 | 10%(day52) | 50%(day52) | Decrease | Significantly decrease | [95] |
| HCC | c-Met/PD-L1 | HepG2-fLuc | 4 | 40%(day50) | 80%(day50) | Decrease | Significantly decrease | [96] |
| HCC | GPC3/ASGR1 | Huh-7/MHCC-97L | 4 | / | / | No Change | Significantly decrease | [97] |
| EOC | Muc16/ WT1 | SKOV3/A2+/GFP+ | 4 | 20%(day60) | 50%(day60) | Decrease | Significantly decrease | [98] |
| EOC | MUC16/ PDL-1 | OVCAR3-MUC16GFP-PDL1-luc | 4 | 48%(day40) | 100%(day40) | No change | Significantly decrease | [99] |
| OC/CC | NKG2D / PDL-1 | HCT116-Luc/ SKOV3-Luc | 4 | 0%(day60) | 100%(day60) | No change | Significantly decrease | [100] |
| OC | TAG-72 /CD47 | TAG-72high OVCAR-3/ TAG-72low MESOV | 4 | / | / | No change | Decrease | [101] |
| GBM | IL-13Rα2/TGF-β | GS001-NSG/C57BL6 | 5 | 20%(day100) | 75%(day100) | Decrease | Significantly decrease | [102] |
| GBM | EGFRvIII /IL-13Rα2 | U87MG-NSG | 4 | 68%(day50) | 100%(day50) | Decrease | Significantly decrease | [20] |
| GBM | EGFR/GD2 | U251 Luc-NXG | 5 | / | / | Decrease | Significantly decrease | [103] |
| iNFPAs | CD87/CD3; CD87/IL-12 | iNFPA PDXs | 5 | 20%(day55) | 90%(day55) | Decrease | Significantly decrease | [104] |
| Breast/lung/ colorectal cancer | TGF-β/ PDL-1 | HCT116- Hsd | 5 | 0%(day53) | 50%(day53) | No change | Significantly decrease | [105] |
| RMS | CD276/FGFR4 | iRFP720+fLuc+ RMS | 5 | 60%(day40) | 100%(day40) | Decrease | Significantly decrease | [106] |
| Solid tumors | CD19/BCMA | HEp-2 / PC-3 /ECA109-NCG | 4 | / | / | Decrease | Significantly decrease | [107] |
| Solid tumors | c-Met/PD-1 | MKN45/A549-NOD/SCID | 5 | 40%(day40) | 80%(day40) | Decrease | Significantly decrease | [108] |
| Pancreatic cancer | CEA/ MSLN | AsPC-1/HT29/U87/PANC-1 | 6 | / | / | Decrease | Significantly decrease | [109] |
Notes: HCC, Hepatocellular Carcinoma; EOC, Epithelial Ovarian Cancer; OC, Ovarian Cancer; CC, Colon Cancer; GBM, Glioblastoma Multiforme; iNFPAs: invasive nonfunctioning pituitary adenomas; RMS, Rhabdomyosarcoma.
Applications of Dual CAR in hematologic tumors
Multiple myeloma
Multiple Myeloma (MM), which accounts for approximately 10% of hematological malignancies, is characterized by abnormal proliferation of plasma cells in the bone marrow, often accompanied by end-organ damage such as acute kidney injury, anemia, destructive osseous bone lesions, and hypercalcemia [50]. Despite the widespread use of novel therapeutic agents, including proteasome inhibitors, monoclonal antibodies and immunomodulatory factors, to improve survival outcomes, the relapse rate remains high, and prognosis is poor due to severe chemoresistance [51]. Currently, two anti-BCMA CAR-T cell products have been approved by the US Food and Drug Administration (FDA) for the treatment of relapsed or refractory (R/R) MM. While BCMA-targeted CAR-T cell therapy has shown high initial response rates, its clinical efficacy is often limited by the temporary nature of responses and frequent relapses, with a median progression-free survival of only 12.2 months, underscoring challenges in achieving long-term curative outcomes due to factors such as CAR-T cell persistence, antigen escape, and the hostile tumor microenvironment [52]. In light of these challenges, emerging preclinical and clinical strategies involving dual-targeted CAR-T cells are being explored to address the limitations of current MM treatment.
G-protein-coupled receptor family C group 5 member D (GPRC5D), a 7-pass transmembrane receptor protein encoded by the GPRC5D gene, has been identified as a potential target to prevent BCMA escape-mediated MM relapse. Single-cell whole-genome sequencing has helped uncover this target, and an FDA-approved antibody targeting GPRC5D (talquetamab) has shown promising response rates in a phase I clinical study, albeit with a distinctive side effect profile [53]. This has led to the exploration of CAR-T designs targeting both BCMA and GPRC5D. Three structural approaches have been investigated; (1) pooled production of single-target CAR-T cells, (2) two distinct CARs from a single vector with bicistronic elements, and (3) the dual-scFv "single-stalk" CAR design. Among these, pooled single CAR-T and bicistronic CAR-T cells exhibited the highest efficacy against BCMA-negative disease. Notably, the bicistronic design proved more effective for diseases co-expressing both BCMA and GPRC5D, highlighting the enhanced therapeutic efficacy of dual-targeted CAR-T cells through intensified interaction with tumor cells [10]. Another potential cause of relapse in MM could be the high expression of CD19 on residual plasma cells, which may drive myeloma propagation and chemotherapy resistance [54]. A novel tandem bispecific CAR targeting both CD19 and BCMA has been shown to induce cytotoxic effects in vitro and tumor regression in xenograft model. This approach resulted in higher anti-tumor efficacy and reduced subsequent recurrence compared to conventional single scFv-CAR-T cells [55]. Recently, an open-label, single-arm phase I/II clinical trial (ChiCTR2000033567) enrolled 50 MM patients, who were treated with BCMA/CD19 dual-targeted CAR-T cells. Of the 46 patients who achieved an overall response (92%), 27 patients with partial remission (PR) or better presented sustained responses, with a 1-year progression-free survival (PFS) rate of 55% during an 11-month follow-up [44]. Additionally, considering that minimal residual myeloma cells express stem-like genes such as CD24, Sun et al. [56] constructed a bispecific BCMA-CD24-CAR-T cell therapy and revealed that this dual-targeted CAR-T cells exhibited increased cytolytic activity and prolonged survival in xenograft models compared to monospecific anti-BCMA CAR-T treatment. Furthermore, several combination targets for CAR-T therapy in MM are under investigation, building on the success of bispecific antibodies, including BCMA/CD3, GPRC5D/CD3 and BCMA/CD19 [57].
B-cell malignant tumor
B-cell malignancies, primarily B-cell lymphomas (BCL), include B-cell Hodgkin's lymphoma and B-cell non-Hodgkin's lymphoma (B-NHL). The typical clinical manifestations of B-cell malignancies include nausea, vomiting, and headaches, often due to elevated intracranial pressure [58]. Diffuse large B-cell lymphoma (DLBCL) is the most common subtype of B-NHL, with a median age at diagnosis of 66 years. In the United States and Western Europe, the number of new DLBCL cases is expected to increase from 29,108 in 2020 to 32,443 in 2025 [59]. Despite recent advancements in clinical treatments, 30% to 40% of patients ultimately succumb to severe complications. Currently, three CAR-T cell products—Axicabtagene ciloleucel (axi-cel), lisocabtagene maraleucel (liso-cel), and tisagenlecleucel (tisa-cel)—have been approved for R/R LBCL patients who have received three or more prior lines of therapy [60]. While CAR-T therapy demonstrates curative potential, limited accessibility and stringent eligibility criteria restrict the number of patients who can benefit from this treatment.
The development of CD19 CAR-T cell therapy has provided significant treatment options for B-cell malignancies, improving survival rates and reducing side effects. However, the loss or mutation of CD19 antigen epitopes remains a major cause of disease relapse [61, 62]. CD22, a member of the sialic acid-binding immunoglobulin-like lectin family, is highly expressed in most B-cell malignancies and has been identified as a potential target to synergize with CD19 in CAR-T cell therapy to prevent cancer recurrence [63]. In this context, four different structures of CD19/CD22 dual-targeted CAR-T cells, with varying linkers and antibody sequences, were designed and compared. The bispecific CAR-T cells with an EAAAK linker exhibited superior pharmacological effects, including enhanced cytotoxicity and higher levels of cytokine secretion, compared to those with a G4S linker. Additionally, dual-targeting or sequential administration of CD19/22 CAR-T cell therapies has been explored to overcome relapses caused by CD19-negative tumor cells in B-NHL patients. However, most patients fail to achieve durable responses, partly due to CAR-T cell exhaustion driven by the PD-1/PD-L1 pathway. To address this issue, a prospective clinical trial combining dual-targeting CD19/22 CAR-T cells with the anti-PD-1 antibody (tislelizumab) was conducted for treating R/R B-NHL [64]. The results indicated that this combination therapy induced safe and durable responses, significantly improving patient prognosis. Epcoritamab and Glofitamab, two bispecific antibodies for CD3 and CD20, were approved by the FDA in May and June of 2023, respectively, for the treatment of DLBCL. In a phase I/II clinical trial involving R/R NHL, 22 DLBCL patients received a full dose of Epcoritamab, with 15 (68%) achieving a positive response. Among these, 10 (45%) patients had a median follow-up of 9.3 months. The ORR was 75%, with a CR rate of 69%. Epcoritamab also induced remissions in patients with aggressive diseases refractory to first-line and/or last-line treatments, with no grade 3 toxicities reported, further supporting its safety profile [65]. Similarly, Glofitamab, with its divalent CD20-targeting structure, shown a higher affinity for its antigen. In a phase I study involving 177 NHL patients, 53% of patients treated with Glofitamab exhibited an ORR, with a CR rate of 36.8%. Among complete responders, 78% sustained their CR at 12 months during a median follow-up of 12.6 months [40]. In recent years, numerous clinical trials have investigated dual-targeted CAR-T therapies in BCL, including various structural designs for dual targets and drug combination therapies, primarily with PD-1 inhibitors [19, 42, 66]. Furthermore, innovative dual-target strategies have been explored for more effective treatments. For instance, CD19/CD20 dual-targeted CAR-T cells have been designed to treat both wild-type BCL and CD19-negative mutants, as well as B-NHL [67, 68]; CD22/CD20 dual-target CAR-T cells demonstrated potent, durable, and dose-dependent activity in vitro and in vivo against primary B-NHL [69]. Additionally, combining CD79b/CD3 bispecific antibodies (bsAbs) with CD19 CAR-T cells offers a promising clinical strategy [70]. Other combinations, such as CD19/CD79a and CD3/CD20, are also being explored for the treatment of R/R BCL [43, 71].
Acute leukemia
Acute leukemia is classified into two main subtypes: acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML). ALL is primarily characterized by either T-lineage or B-lineage involvement and is considered the most malignant in childhood [72]. AML, on the other hand, is a rapidly progressive hematologic malignancy characterized by the clonal expansion and abnormal function of immature myeloid precursors. Chemotherapy and hematopoietic stem cell transplantation are two common treatment strategies for leukemia [73]; however, the high rates of complications post-transplantation and a low 5-year survival rate seriously impact the overall therapeutic effect [74]. Although CAR-T cell therapy has been utilized in the treatment of acute leukemia, challenges such as tumor antigen escape, severe CRS, and relapse after treatment are still observed in some patients [75], highlighting the need for novel therapeutic approaches.
Since the approval of Blinatumomab as the first bispecific antibody for B-ALL in 2017 [49], numerous studies targeting dual antigens in leukemia have been initiated. One of the key challenges in CAR-T therapy for ALL is CD19-negative relapse, which remains a primary cause of treatment failure [76]. To address this, a clinical study targeting CD19-negative NALM6 with CD19/CD22 dual-targeted CAR-T cells showed promising results, demonstrating long-lasting efficacy [46]. In detail, the combination of CD19 and a novel CD22 CAR exhibited effective cytotoxicity even at low antigen densities. Twelve patients with advanced B-ALL were enrolled, and of these, 10 cases (83%) achieved measurable residual disease (MRD)-negative complete remission two months post-infusion. Furthermore, with a median follow-up of 8.7 months, none of the patients relapsed due to antigen-negative escape. The overall survival and event-free survival rates at 6 and 12 months were 75% and 60%, respectively. To date, a series of preclinical studies and clinical trials involving dual-targeting CD19/CD22 are actively being conducted in leukemia [47, 48, 77]. C-type lectin-like molecule 1 (CLL-1) is a transmembrane glycoprotein, and CD123, another transmembrane glycoprotein, is part of the interleukin-3 (IL-3) receptor alpha chain. Previous studies have shown that leukemic stem cells (LSCs) play a critical role in leukemia onset and relapse. Both CD123 and CLL-1 are highly expressed on the surface of most leukemia cells, including LSCs, making them promising therapeutic targets for AML [78-80]. For instance, Wang et al. [81] developed tandem CAR-T cells targeting CLL-1 and CD123 to assess their therapeutic potential in AML in vitro, and they found that these dual-targeting CAR-T cells exhibited robust killing effects and released a large number of cytokines, demonstrating a significant ability to kill single antigens and multi-target tumour cells. CD33, as a myeloid differentiation antigen, is also highly expressed on the blasts and LSCs in AML patients, while it is almost absent from normal hematopoietic stem cells [82]. Clinical therapies targeting CD33 can effectively eliminate the transformed clone, they often fail to eradicate the precursor LSCs, which can lead to disease relapse [83]. IL-10 has been shown to enhance the stemness of AML cells through various signaling pathways, and CAR-T cells targeting IL-10R have demonstrated cytotoxic effects against AML cells. A recent study developed anti-IL10R CAR-T cells that secrete CD33-targeting bsAbs, aimed at combating tumor heterogeneity and eradicating both LSCs and AML blasts. Moreover, these CAR-T cells, which deliver bsAbs directly to the tumor sites, can help overcome pharmacokinetic challenges and improve therapeutic efficacy [84]. In addition, several other dual-target strategies have emerged in the past two years to address antigen escape in various forms of leukemias [85-89]. Notable dual-target combinations include NKG2D/PD‑L1, IL-3 -zetakine /CD33, CD33/CD146, CD19/BAFF-R, and CD123/NKG2DLs. TCRs represent another promising approach for targeting tumor-derived neoantigens, as they can reduce the risk of off-tumor effects. TCR-based therapies have been considered highly specific alternatives for treating leukemia [90]. For example, Teppert et al. [91] designed CAR'TCR-T cells co-expressing dNPM1-TCR and CD33-CAR for AML treatment. This strategy significantly enhanced anti-tumor cytotoxicity, demonstrating the potential of co-expressing both CAR and transgenic TCRs within a single T cell. Furthermore, ongoing studies are investigating the antibody-TCR dual-target approach, combining Wilms tumor 1 protein (WT1) and CD33, which is showing promise in early trials [92]. These developments suggest that integrating TCRs into CAR-T cell therapies may open up new therapeutic avenues for AML and other malignancies.
Applications of dual-targeted CAR-T in solid tumors
Hepatocellular carcinoma
Primary liver cancer is a global health issue, ranking as the sixth most common cancer worldwide and the fourth leading cause of cancer-related mortality. Hepatocellular carcinoma (HCC), accounting for 75-80% of liver cancers, is the predominant histological type [110, 111]. The etiology of HCC is primarily rooted in chronic hepatitis, hepatitis B virus (HBV)/hepatitis C virus (HCV)-induced cirrhosis, alcoholic cirrhosis, dietary aflatoxin exposure, non-alcoholic steatohepatitis, alpha-1-antitrypsin deficiency, and hemochromatosis [112]. Current clinical treatment options for HCC include surgical intervention, CAR-T cell therapy, immune checkpoint inhibitors (ICIs), tyrosine kinase inhibitors (TKIs), and antibody therapies. However, HCC remains largely incurable due to tumor heterogeneity and metastasis [113, 114]. Recent research on immunotherapies employing dual-targeting strategies offers a promising new approach to overcoming the challenges of current treatments. Glypican 3 (GPC3), a tumor-associated antigen, is highly expressed in over 70% of HCC cases, but is strictly suppressed in normal liver tissue [115, 116]. Fibroblast activation protein (FAP) is a type-II transmembrane serine protease secreted from cancer-associated fibroblasts (CAFs). During liver carcinogenesis, FAP promotes fibrosis in response to early liver injury, then facilitates tumor cell proliferation, and contributes to immune suppression [117-119]. While CAR-T cells targeting GPC3 alone have shown some therapeutic efficacy, their clinical use is limited by several challenges [120]. In response, bispecific CAR-T cells incorporating tandem scFvs targeting both FAP and GPC3 scFv have been developed to recognize and eliminate tumor cells expressing either or both antigens [39]. Both in vitro and in vivo studies have shown that these dual-targeted CAR-T cells significantly enhance therapeutic efficacy against HCC, particularly in tumors with high expression of GPC3 or FAP. These cells suppress tumor growth and prolong survival in tumor-bearing mice, highlighting their potential to prevent antigen escape and combat heterogeneous HCC. ICIs, such as anti PD-1/PD-L1 monoclonal antibodies, have shown durable responses in a subset of HCC patients, particularly those with high PD-L1 expression or tumor mutational burden, however, the response rates in HCC are generally low (10-20%), and acquired resistance is common due to compensatory upregulation of alternative immune checkpoints (e.g., CTLA-4, TIM-3) [121, 122]. To address this, a recent study developed a dual-targeting CAR-T cell (GPC3/PD-1) that recognizes GPC3 combining antigen-specific targeting PD-1 to block immune checkpoint, potentially overcoming ICI resistance by directly targeting tumor cells while modulating the TME. This strategy demonstrated greater resistance to PD-1/PD-L1 pathway inhibition, characterized by reduced inhibitory receptor expression and a less differentiated phenotype, resulting in more potent anti-tumor activity compared to single-target CAR-T cells [93]. Epidermal Growth Factor Receptor (EGFR), a receptor tyrosine kinase (RTK) of the ErbB family, is highly expressed in human HCC and is associated with more aggressive tumor growth. It is also expressed at lower levels in liver epithelial cells, [123, 124]. Combining GPC3 with EGFR, third-generation GPC3-EGFR CAR-T cells have been designed, showing enhanced proliferation and cytotoxicity while minimizing non-tumor toxicity [95]. Furthermore, Chen et al. [97] constructed CAR-T cells carrying complementary CARs against GPC3 and ASGR1 (a liver tissue-specific protein). These dual-targeted CAR-T cells reduced the risk of on-target, off-tumor toxicity while maintaining anti-tumor activity in dual-positive HCC. In addition to these approaches, other dual-targeting strategies for HCC treatment are being explored to address antigen escape and the challenges posed by tumor microenvironment, such as targeting c-Met/PD-L1 and CD133/GPC3 [94, 96].
Ovarian cancer
Ovarian cancer (OC) is the most lethal gynecological malignancy, with a poor clinical prognosis due to its high metastatic potential, drug resistance, and the lack of early detection and screening technologies [125]. In addition to standard treatments such as surgery, radiotherapy, chemotherapy, and targeted therapies, cell immunotherapy, particularly CAR-T cell therapy, has emerged as an effective approach for cancer treatment [126, 127]. MUC16 (also known as CA125) is a cell surface mucin that is highly expressed in epithelial ovarian tumors, making it a prominent marker in OC development [128]. Recent findings have shown that MUC16 can suppress antitumor activity of immune cells, facilitating immune evasion by tumor cells [129, 130]. Furthermore, Wilms tumor 1 (WT1), an intracellular transcription factor, is commonly overexpressed in various hematological and solid cancers including OC [131]. To overcome the challenge of low MUC16 expression on ovarian cancer cells, MUC16-specific CAR-T cells were engineered to secrete a bispecific T cell engager that targets WT1, enabling the CAR-T cells to kill ovarian cancer cells via an orthogonal mechanism. This dual-target approach addresses tumor heterogeneity and enhances therapeutic efficacy by utilizing two distinct killing mechanisms [98]. This study is the first to demonstrate the combination of targeting both intracellular and extracellular antigens, reducing the risk of antigen escape and improving overall treatment effectiveness. NKG2D is an activating receptor on NK cells that plays a crucial role in mediating immune cell activation and the destruction of target cells [132]. Previous research has shown that over 80% of human ovarian cancer ascites samples express NKG2D ligands on their surface, and various NKG2D ligands are also found in human ovarian cancer cell lines [133, 134]. PD-1 is another key target in cancer immunotherapy. Given the widespread expression of NKG2D and PD-1 ligands in various human cancers, these factors have been considered as promising targets for cancer treatment. Jiang et al. [100] combined a first-generation CAR targeting NKG2D ligands with a CAR targeting PD-1 ligands, generating a novel dual CAR that demonstrated broad clinical potential in precision cancer immunotherapy. Moreover, a tandem PD1-antiMUC16 dual CAR-T cell therapy has been developed. Data from preclinical studies showed that these dual CAR-T cells significantly enhanced the cytotoxicity against ovarian cancer OVCAR-3 cells and extended the survival time of tumor-bearing mice. This dual-targeted CAR-T approach exhibited more potent antitumor activity in vivo compared to single CAR-T cell therapies [99]. Other dual-targeting strategies, such as targeting TAG-72/CD47 [101], are also under investigation. These dual-target approaches have the potential to effectively prevent immune escape in OC and address challenges posed the tumor microenvironment, improving upon the limitations of single-target CAT therapies.
Glioblastoma multiforme
Glioblastoma multiforme (GBM) is the most common and aggressive form of primary malignant brain tumor, representing the highest grade of astrocytoma [135]. GBM is characterized by its high invasiveness and resistance to almost all therapeutic interventions, including the combination of chemotherapy and radiotherapy following surgical resection. The challenges in treating GBM primarily arise from the drug resistance of malignant glioblastoma cells, as well as the complex distribution of inter- and intra-tumoral heterogeneity. As a result, the 5-year overall survival rate remains below 10% after treatment [136, 137]. While current research on single-target antigens has shown promising therapeutic effects, complete and durable responses are rare, making it difficult to achieve robust efficacy [138]. Therefore, developing dual-targeting strategies has become essential to address these challenges.
The epidermal growth factor receptor variant III (EGFRvIII) and the interleukin-13 receptor alpha 2 (IL-13Rα2) are specially expressed on the surface of GBM cells [139], yet are either completely absent or minimally expressed in normal somatic tissues. Given that EGFRvIII and IL-13Rα2 are co-expressed in the same tumor cells, the intracellular tandem specificity between these two molecules may confer a growth advantage to the tumor, making them an ideal combination for simultaneous targeting [140]. A recent study developed a novel bispecific tandem CAR-T (TanCART) cell capable of targeting both EGFRvIII and IL-13Rα2. The results demonstrated that TanCAR-T cells exhibited enhanced activity and potent cytotoxicity, achieving complete and durable tumor responses in a heterogeneous GBM mouse model [20]. This study highlights the effectiveness of TanCART in targeting heterogeneous brain tumors and provides further evidence supporting the development of multispecific CAR-T cell therapies for GBM. Transforming growth factor-beta (TGF-β) is overexpressed in gliomas and plays a critical role in maintaining the GBM tumor microenvironment by promoting the tumorigenicity of glioma-initiating stem cells, as well as tumor cell proliferation, invasiveness, and immune evasion [141, 142]. A recent study designed a single-chain bispecific CAR targeting IL-13Rα2 and TGF-β. This CAR programs tumor-specific T cells to convert TGF-β from an immunosuppressive agent into an immunostimulatory one, thereby reshaping the immunosuppressive TME and enhancing antitumor responses in GBM [102]. Treatment with IL-13Rα2/TGF-β CAR-T cells in human and mouse GBM models has demonstrated increased T cell infiltration, reduced levels of suppressive myeloid cells in the tumor-bearing brain, and improved survival rates in patient-derived GBM xenografts and syngeneic mouse models. This study offers a promising and novel therapeutic approach for the clinical translation of bispecific IL13Rα2/TGF-β CAR-T cells to overcome the immunosuppressive TME in GBM. Additionally, Saleh et al. generated [103] RevCAR T cells targeting both EGFR and disialoganglioside (GD2), which marked the first successful application of RevCAR T cells in a dual-targeting approach to efficiently, specifically, and programmably eliminate GBM cells both in vitro and in vivo.
Other solid tumors
In addition to extensive research on solid tumors, there has been a growing focus in recent years on dual-targeting approaches for various types of solid tumors. For instance, CD87×CD3 BiTE antibodies and CD87/IL-12 CAR-T cells have been designed to target non-functioning pituitary adenomas (NFPA) [104]. The CD87×CD3 BiTE antibody effectively reduces tumor cell proliferation, demonstrating significant lytic activity both in tumor cells and in preclinical models. Furthermore, CD87/IL-12 CAR-T cells exhibited enhanced antitumor activity, inducing tumor regression more effectively than CD87 single-target CAR-T cells in both in vivo studies and three-dimensional co-culture models. Moreover, bispecific therapies targeting TGF-β and PD-L1 in various tumors, including lung, breast, and colorectal cancers, have been shown to significantly enhance T cell activation and cytotoxic responses [105, 143, 144]. Dual-functional CAR-T cells targeting c-Met and PD-1, with PD-1 blocking capability, significantly boosted the cytotoxicity of CAR-T cells, demonstrating a stronger ability to inhibit tumor growth and prolong the survival of tumor-bearing mice in gastric cancer models [108]. Dual CAR-T cells targeting CD276/FGFR4 effectively killed of rhabdomyosarcoma (RMS) cells in vitro and eradicated in situ RMS in preclinical models [106]. Increasing numbers of Phase I clinical trials are investigating the safety and efficacy of bispecific antibodies targeting PD-1 and LAG-3 across various cancers, including epithelial ovarian cancer (EOC), triple-negative breast cancer (TNBC), non-small cell lung cancer (NSCLC), small cell lung cancer (SCLC), cervical cancer, and cholangiocarcinoma [145]. Moreover, patients with refractory primary central nervous system lymphoma (PCNSL) have achieved long-term complete remission following treatment with CD19/CD22 dual-targeting CAR-T cells in combination with PD-1 and BTK inhibitors [146]. CAR-T cells targeting CD30 and carcinoembryonic antigen (CEA) have shown improved redirected immune responses against colorectal cancer [21]. Furthermore, CAR-T cells targeting both CEA and mesothelin (MSLN) exhibit potent antitumor activity in pancreatic cancer, significantly inhibiting tumor cell growth without affecting normal tissues [109]. These examples highlight the increasing momentum of dual-targeting studies across a wide range of solid tumors. In summary, dual-targeting CAR-T cells demonstrate enhanced persistence within tumor tissues, reduced expression of inhibitory receptors, and a less differentiated phenotype, thereby achieving a more potent and sustained antitumor effect.
Discussion and Prospects
Despite the promising preclinical and clinical outcomes of dual CAR-T cell therapy for both hematological and solid tumors, several challenges remain before its widespread clinical application. This section discusses the current limitations of dual CAR-T cell therapy, potential strategies to address these challenges, and future directions for development.
First, the complexity of designing the dual CAR structure of CAR-T cells poses a major challenge. Developing appropriate vector systems is essential for enhancing the functionality of CAR-T cells [147]. The rational design of two CAR domains is critical to avoid internal competition between the domains and to ensure efficient dual-specific recognition. In addition, the high manufacturing costs and complex production processes of dual CAR-T cells may limit their accessibility, particularly in low- and middle-income countries [16, 148]. Second, the safety profile of dual-targeted CAR-T cells is a critical consideration for the clinical translation. In a clinical trial for non-Hodgkin lymphoma, tandem CD19/CD20 CAR-T cells achieved a median PFS of 23.9 months, significantly longer than single-target CAR-T therapies (8.9 months), with no increased incidence of CRS or neurotoxicity [149]. Although dual-targeted CAR-T cells can mitigate on-target/off-tumor toxicity by targeting tumor-specific antigen pairs, they face unique challenges including TME-mediated exhaustion and and cytokine storm risk [11]. Emerging strategies address these risks through engineering innovations, such as logic-gated activation, metabolic reprogramming, and epigenetic modulation [150]. For instance, AND-gated EGFRvIII/IL13Rα2) significantly reduced uncontrolled cytokine release [151]. IL-6 receptor blockade (tocilizumab) combined with short-course corticosteroids remains first-line therapy, resolving grade 3-4 CRS in 80% of cases [152]. Moreover, decitabine-primed tandem CD19/CD22 dual-targeted CAR-T therapy maintained complete remission for a 35-month follow-up period without neurotoxicity [146]. Also, the FDA emphasizes key insights and recommendations for optimizing safety, including safety switches, dose optimization, and biomarker monitoring, as highlighted in the FDA's guidance document Considerations for the Development of Chimeric Antigen Receptor (CAR) T Cell Products. Furthermore, the specificity and efficacy of dual-targeted CAR-T cells across diverse tumor types require validation through large-scale clinical trials with extended follow-up periods [153]. To enhance therapeutic outcomes, strategic approaches can be prioritized, such as optimizing co-stimulatory signals in CAR-T cells (e.g., combining CD28 and 4-1BB domains), improving the tumor microenvironment (e.g., immunosuppressive cell depletion, physical barrier disruption, hypoxia and metabolic stress alleviation), and exploring novel dual CAR-T cell designs (e.g., logic-gated CARs) to address antigen heterogeneity and enhance persistence [11, 28, 154, 155].
In recent years, advancements in gene-editing technologies (e.g., CRISPR-Cas9) and a deeper understanding of the tumor immune microenvironment have paved the way for more personalized and precise treatments using dual CAR-T cells, enabling enhanced targeting of heterogeneous tumors and overcoming immune evasion mechanisms [156, 157]. Moreover, combination therapies incorporating immune checkpoint inhibitors, oncolytic viruses, or other immunomodulatory agents may further enhance the therapeutic efficacy of dual-targeted CAR-T cells, offering new hope for cancer patients. As clinical research continues to progress, dual-targeted CAR-T cell therapy is expected to become a significant modality in cancer treatment in the near future.
Supplementary Material
Supplementary table.
Acknowledgements
Funding
This study was supported by Wuhan East Lake High-tech Zone “JieBangGuaShuai” Project (2022KJB113), Open Project Funding of the Key Laboratory of Fermentation Engineering (Ministry of Education) (202409FE06), Hubei Provincial Natural Science Foundation of China (2024AFB793), Guangdong Basic and Applied Basic Research Foundation (2021A1515011272), Graduate Innovation and Entrepreneurship Project of Wuhan University of Science and Technology (JCX2023064).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229-63
2. Albano D, Benenati M, Bruno A, Bruno F, Calandri M, Caruso D. et al. Imaging side effects and complications of chemotherapy and radiation therapy: a pictorial review from head to toe. Insights Imaging. 2021;12:76
3. Kaur R, Bhardwaj A, Gupta S. Cancer treatment therapies: traditional to modern approaches to combat cancers. Mol Biol Rep. 2023;50:9663-76
4. Rui R, Zhou L, He S. Cancer immunotherapies: advances and bottlenecks. Front Immunol. 2023;14:1212476
5. Saleh K, Pasquier F, Bigenwald C, De Botton S, Ribrag V, Castilla-Llorente C. CAR T-Cells for the Treatment of B-Cell Acute Lymphoblastic Leukemia. J Clin Med. 2023;12:6883
6. Ernst M, Oeser A, Besiroglu B, Caro-Valenzuela J, Abd El Aziz M, Monsef I. et al. Chimeric antigen receptor (CAR) T-cell therapy for people with relapsed or refractory diffuse large B-cell lymphoma. Cochrane Database Syst Rev. 2021;9:Cd013365
7. Haslauer T, Greil R, Zaborsky N, Geisberger R. CAR T-Cell Therapy in Hematological Malignancies. Int J Mol Sci. 2021;22:8996
8. Maalej KM, Merhi M, Inchakalody VP, Mestiri S, Alam M, Maccalli C. et al. CAR-cell therapy in the era of solid tumor treatment: current challenges and emerging therapeutic advances. Mol Cancer. 2023;22:20
9. Roddie C, Lekakis LJ, Marzolini MAV, Ramakrishnan A, Zhang Y, Hu Y. et al. Dual targeting of CD19 and CD22 with bicistronic CAR-T cells in patients with relapsed/refractory large B-cell lymphoma. Blood. 2023;141:2470-82
10. Fernández de Larrea C, Staehr M, Lopez AV, Ng KY, Chen Y, Godfrey WD. et al. Defining an Optimal Dual-Targeted CAR T-cell Therapy Approach Simultaneously Targeting BCMA and GPRC5D to Prevent BCMA Escape-Driven Relapse in Multiple Myeloma. Blood Cancer Discov. 2020;1:146-54
11. Hirabayashi K, Du H, Xu Y, Shou P, Zhou X, Fucá G. et al. Dual Targeting CAR-T Cells with Optimal Costimulation and Metabolic Fitness enhance Antitumor Activity and Prevent Escape in Solid Tumors. Nat Cancer. 2021;2:904-18
12. Ruella M, Maus MV. Catch me if you can: Leukemia Escape after CD19-Directed T Cell Immunotherapies. Comput Struct Biotechnol J. 2016;14:357-62
13. Majzner RG, Mackall CL. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018;8:1219-26
14. Dagar G, Gupta A, Masoodi T, Nisar S, Merhi M, Hashem S. et al. Harnessing the potential of CAR-T cell therapy: progress, challenges, and future directions in hematological and solid tumor treatments. J Transl Med. 2023;21:449
15. Jogalekar MP, Rajendran RL, Khan F, Dmello C, Gangadaran P, Ahn BC. CAR T-Cell-Based gene therapy for cancers: new perspectives, challenges, and clinical developments. Front Immunol. 2022;13:925985
16. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11:69
17. Zhang X, Zhu L, Zhang H, Chen S, Xiao Y. CAR-T Cell Therapy in Hematological Malignancies: Current Opportunities and Challenges. Front Immunol. 2022;13:927153
18. Chen Z, Liu Y, Chen N, Xing H, Tian Z, Tang K. et al. Loop CD20/CD19 CAR-T cells eradicate B-cell malignancies efficiently. Sci China Life Sci. 2023;66:754-70
19. Wei G, Zhang Y, Zhao H, Wang Y, Liu Y, Liang B. et al. CD19/CD22 Dual-Targeted CAR T-cell Therapy for Relapsed/Refractory Aggressive B-cell Lymphoma: A Safety and Efficacy Study. Cancer Immunol Res. 2021;9:1061-70
20. Schmidts A, Srivastava AA, Ramapriyan R, Bailey SR, Bouffard AA, Cahill DP. et al. Tandem chimeric antigen receptor (CAR) T cells targeting EGFRvIII and IL-13Rα2 are effective against heterogeneous glioblastoma. Neurooncol Adv. 2023;5:vdac185
21. Hombach AA, Rappl G, Abken H. Blocking CD30 on T Cells by a Dual Specific CAR for CD30 and Colon Cancer Antigens Improves the CAR T Cell Response against CD30(-) Tumors. Mol Ther. 2019;27:1825-35
22. Dai Z, Mu W, Zhao Y, Cheng J, Lin H, Ouyang K. et al. T cells expressing CD5/CD7 bispecific chimeric antigen receptors with fully human heavy-chain-only domains mitigate tumor antigen escape. Signal Transduct Target Ther. 2022;7:85
23. Feng Y, Liu X, Li X, Zhou Y, Song Z, Zhang J. et al. Novel BCMA-OR-CD38 tandem-dual chimeric antigen receptor T cells robustly control multiple myeloma. Oncoimmunology. 2021;10:1959102
24. Dai Q, Han P, Qi X, Li F, Li M, Fan L. et al. 4-1BB Signaling Boosts the Anti-Tumor Activity of CD28-Incorporated 2(nd) Generation Chimeric Antigen Receptor-Modified T Cells. Front Immunol. 2020;11:539654
25. Bôle-Richard E, Fredon M, Biichlé S, Anna F, Certoux JM, Renosi F. et al. CD28/4-1BB CD123 CAR T cells in blastic plasmacytoid dendritic cell neoplasm. Leukemia. 2020;34:3228-41
26. Larcombe-Young D, Whilding L, Davies DM, Draper B, Bechman N, Maher J. Generation of human parallel chimeric antigen receptor (pCAR) T cells to achieve synergistic T cell co-stimulation. STAR Protoc. 2022;3:101414
27. Zhao W, Yao Y, Li Q, Xue Y, Gao X, Liu X. et al. Molecular mechanism of co-stimulatory domains in promoting CAR-T cell anti-tumor efficacy. Biochem Pharmacol. 2024;227:116439
28. Honikel MM, Olejniczak SH. Co-Stimulatory Receptor Signaling in CAR-T Cells. Biomolecules. 2022;12:1303
29. Katsarou A, Sjöstrand M, Naik J, Mansilla-Soto J, Kefala D, Kladis G. et al. Combining a CAR and a chimeric costimulatory receptor enhances T cell sensitivity to low antigen density and promotes persistence. Sci Transl Med. 2021;13:eabh1962
30. Li N, Quan A, Li D, Pan J, Ren H. The IgG4 hinge with CD28 transmembrane domain improves V(H)H-based CAR T cells targeting a membrane-distal epitope of GPC1 in pancreatic cancer. Nat Commun. 2023;14:1986
31. Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA. et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med. 2021;13:eabe7378
32. Cho JH, Okuma A, Al-Rubaye D, Intisar E, Junghans RP, Wong WW. Engineering Axl specific CAR and SynNotch receptor for cancer therapy. Sci Rep. 2018;8:3846
33. Fu Y, Wang T, Ronald JA. A synthetic notch (synNotch) system linking intratumoral immune-cancer cell communication to a synthetic blood biomarker assay. Front Pharmacol. 2023;14:1304194
34. Yang M, Tang X, Zhang Z, Gu L, Wei H, Zhao S. et al. Tandem CAR-T cells targeting CD70 and B7-H3 exhibit potent preclinical activity against multiple solid tumors. Theranostics. 2020;10:7622-34
35. Muliaditan T, Halim L, Whilding LM, Draper B, Achkova DY, Kausar F. et al. Synergistic T cell signaling by 41BB and CD28 is optimally achieved by membrane proximal positioning within parallel chimeric antigen receptors. Cell Rep Med. 2021;2:100457
36. Requejo Cier CJ, Valentini N, Lamarche C. Unlocking the potential of Tregs: innovations in CAR technology. Front Mol Biosci. 2023;10:1267762
37. Hyrenius-Wittsten A, Su Y, Park M, Garcia JM, Alavi J, Perry N. et al. SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Sci Transl Med. 2021;13:eabd8836
38. Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019;16:372-85
39. Zhou L, Li Y, Zheng D, Zheng Y, Cui Y, Qin L. et al. Bispecific CAR-T cells targeting FAP and GPC3 have the potential to treat hepatocellular carcinoma. Mol Ther Oncol. 2024;32:200817
40. Hutchings M, Morschhauser F, Iacoboni G, Carlo-Stella C, Offner FC, Sureda A. et al. Glofitamab, a Novel, Bivalent CD20-Targeting T-Cell-Engaging Bispecific Antibody, Induces Durable Complete Remissions in Relapsed or Refractory B-Cell Lymphoma: A Phase I Trial. J Clin Oncol. 2021;39:1959-70
41. Spiegel JY, Patel S, Muffly L, Hossain NM, Oak J, Baird JH. et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat Med. 2021;27:1419-31
42. Zhou Y, Li J, Zhang X, Jia T, Zhang B, Dai N. et al. Prognostic Value of Radiomic Features of (18)F-FDG PET/CT in Patients With B-Cell Lymphoma Treated With CD19/CD22 Dual-Targeted Chimeric Antigen Receptor T Cells. Front Oncol. 2022;12:834288
43. Thieblemont C, Phillips T, Ghesquieres H, Cheah CY, Clausen MR, Cunningham D. et al. Epcoritamab, a Novel, Subcutaneous CD3xCD20 Bispecific T-Cell-Engaging Antibody, in Relapsed or Refractory Large B-Cell Lymphoma: Dose Expansion in a Phase I/II Trial. J Clin Oncol. 2023;41:2238-47
44. Shi M, Wang J, Huang H, Liu D, Cheng H, Wang X. et al. Bispecific CAR T cell therapy targeting BCMA and CD19 in relapsed/refractory multiple myeloma: a phase I/II trial. Nat Commun. 2024;15:3371
45. Garfall AL, Cohen AD, Susanibar-Adaniya SP, Hwang WT, Vogl DT, Waxman AJ. et al. Anti-BCMA/CD19 CAR T Cells with Early Immunomodulatory Maintenance for Multiple Myeloma Responding to Initial or Later-Line Therapy. Blood Cancer Discov. 2023;4:118-33
46. Ghorashian S, Lucchini G, Richardson R, Nguyen K, Terris C, Guvenel A. et al. CD19/CD22 targeting with cotransduced CAR T cells to prevent antigen-negative relapse after CAR T-cell therapy for B-cell ALL. Blood. 2024;143:118-23
47. Hu Y, Zhou Y, Zhang M, Ge W, Li Y, Yang L. et al. CRISPR/Cas9-Engineered Universal CD19/CD22 Dual-Targeted CAR-T Cell Therapy for Relapsed/Refractory B-cell Acute Lymphoblastic Leukemia. Clin Cancer Res. 2021;27:2764-72
48. Cordoba S, Onuoha S, Thomas S, Pignataro DS, Hough R, Ghorashian S. et al. CAR T cells with dual targeting of CD19 and CD22 in pediatric and young adult patients with relapsed or refractory B cell acute lymphoblastic leukemia: a phase 1 trial. Nat Med. 2021;27:1797-805
49. Kantarjian H, Stein A, Gökbuget N, Fielding AK, Schuh AC, Ribera JM. et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N Engl J Med. 2017;376:836-47
50. Minnie SA, Hill GR. Immunotherapy of multiple myeloma. J Clin Invest. 2020;130:1565-75
51. Li X, Wang S, Xie Y, Jiang H, Guo J, Wang Y. et al. Deacetylation induced nuclear condensation of HP1γ promotes multiple myeloma drug resistance. Nat Commun. 2023;14:1290
52. Roex G, Timmers M, Wouters K, Campillo-Davo D, Flumens D, Schroyens W. et al. Safety and clinical efficacy of BCMA CAR-T-cell therapy in multiple myeloma. J Hematol Oncol. 2020;13:164
53. Chari A, Minnema MC, Berdeja JG, Oriol A, van de Donk N, Rodríguez-Otero P. et al. Talquetamab, a T-Cell-Redirecting GPRC5D Bispecific Antibody for Multiple Myeloma. N Engl J Med. 2022;387:2232-44
54. Wang Y, Cao J, Gu W, Shi M, Lan J, Yan Z. et al. Long-Term Follow-Up of Combination of B-Cell Maturation Antigen and CD19 Chimeric Antigen Receptor T Cells in Multiple Myeloma. J Clin Oncol. 2022;40:2246-56
55. Kang L, Zhang J, Li M, Xu N, Qi W, Tan J. et al. Characterization of novel dual tandem CD19/BCMA chimeric antigen receptor T cells to potentially treat multiple myeloma. Biomark Res. 2020;8:14
56. Sun F, Cheng Y, Wanchai V, Guo W, Mery D, Xu H. et al. Bispecific BCMA/CD24 CAR-T cells control multiple myeloma growth. Nat Commun. 2024;15:615
57. Hou J, Li Y, Lin Q. Bispecific antibodies and dual-targeting CAR-T cells for multiple myeloma: latest updates from the 2023 ASCO annual meeting. Exp Hematol Oncol. 2023;12:74
58. Pinto SN, Liu CJ, Nelson MD Jr, Bluml S, Livingston D, Tamrazi B. Neuroimaging of complications arising after CD19 chimeric antigen receptor T-cell therapy: A review. J Neuroimaging. 2023;33:703-15
59. Kanas G, Ge W, Quek RGW, Keeven K, Nersesyan K, Jon EA. Epidemiology of diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma (FL) in the United States and Western Europe: population-level projections for 2020-2025. Leuk Lymphoma. 2022;63:54-63
60. Trabolsi A, Arumov A, Schatz JH. Bispecific antibodies and CAR-T cells: dueling immunotherapies for large B-cell lymphomas. Blood Cancer J. 2024;14:27
61. Wang J, Hu Y, Huang H. Acute lymphoblastic leukemia relapse after CD19-targeted chimeric antigen receptor T cell therapy. J Leukoc Biol. 2017;102:1347-56
62. Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M. et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med. 2018;24:1504-6
63. Ma R, You F, Tian S, Zhang T, Tian X, Xiang S. et al. Enhanced efficacy of CD19/CD22 bispecific CAR-T cells with EAAAK linker on B-cell malignancies. Eur J Haematol. 2024;112:64-74
64. Zhang Y, Geng H, Zeng L, Li J, Yang Q, Jia S. et al. Tislelizumab augment the efficacy of CD19/22 dual-targeted chimeric antigen receptor T cell in advanced stage relapsed or refractory B-cell non-Hodgkin lymphoma. Hematol Oncol. 2024;42:e3227
65. Hutchings M, Mous R, Clausen MR, Johnson P, Linton KM, Chamuleau MED. et al. Dose escalation of subcutaneous epcoritamab in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an open-label, phase 1/2 study. Lancet. 2021;398:1157-69
66. Huang C, Zhang HC, Ho JY, Liu RX, Wang L, Kuang N. et al. Dual specific CD19/CD22-targeted chimeric antigen receptor T-cell therapy for refractory diffuse large B-cell lymphoma: A case report. Oncol Lett. 2020;20:21
67. Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol Res. 2016;4:498-508
68. Russler-Germain DA, Ghobadi A. T-cell redirecting therapies for B-cell non-Hodgkin lymphoma: recent progress and future directions. Front Oncol. 2023;13:1168622
69. Aranda-Orgilles B, Chion-Sotinel I, Skinner J, Grudman S, Mumford B, Dixon C. et al. Preclinical Evidence of an Allogeneic Dual CD20xCD22 CAR to Target a Broad Spectrum of Patients with B-cell Malignancies. Cancer Immunol Res. 2023;11:946-61
70. Zheng WW, Zhou H, Li P, Ye SG, Abudureheman T, Yang LT. et al. Anti-CD79b/CD3 bispecific antibody combined with CAR19-T cells for B-cell lymphoma treatment. Cancer Immunol Immunother. 2023;72:3739-53
71. Leung I, Templeton ML, Lo Y, Rajan A, Stull SM, Garrison SM. et al. Compromised antigen binding and signaling interfere with bispecific CD19 and CD79a chimeric antigen receptor function. Blood Adv. 2023;7:2718-30
72. Malard F, Mohty M. Acute lymphoblastic leukaemia. Lancet. 2020;395:1146-62
73. Gratwohl A, Baldomero H, Passweg J. Hematopoietic stem cell transplantation activity in Europe. Curr Opin Hematol. 2013;20:485-93
74. Takami A. Hematopoietic stem cell transplantation for acute myeloid leukemia. Int J Hematol. 2018;107:513-8
75. Wang X, Xiao Q, Wang Z, Feng WL. CAR-T therapy for leukemia: progress and challenges. Transl Res. 2017;182:135-44
76. Dourthe ME, Rabian F, Yakouben K, Chevillon F, Cabannes-Hamy A, Méchinaud F. et al. Determinants of CD19-positive vs CD19-negative relapse after tisagenlecleucel for B-cell acute lymphoblastic leukemia. Leukemia. 2021;35:3383-93
77. Kokalaki E, Ma B, Ferrari M, Grothier T, Hazelton W, Manzoor S. et al. Dual targeting of CD19 and CD22 against B-ALL using a novel high-sensitivity aCD22 CAR. Mol Ther. 2023;31:2089-104
78. Döhner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015;373:1136-52
79. Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, Wilkinson L. et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5:31-42
80. Ehninger A, Kramer M, Röllig C, Thiede C, Bornhäuser M, von Bonin M. et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014;4:e218
81. Wang XY, Bian MR, Lin GQ, Yu L, Zhang YM, Wu DP. Tandem bispecific CD123/CLL-1 CAR-T cells exhibit specific cytolytic effector functions against human acute myeloid leukaemia. Eur J Haematol. 2024;112:83-93
82. Laszlo GS, Estey EH, Walter RB. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev. 2014;28:143-53
83. Walter RB, Appelbaum FR, Estey EH, Bernstein ID. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood. 2012;119:6198-208
84. Yan Z, Gu R, Ma H, Chen N, Zhang T, Xu Y. et al. A dual-targeting approach with anti-IL10R CAR-T cells engineered to release anti-CD33 bispecific antibody in enhancing killing effect on acute myeloid leukemia cells. Cell Oncol (Dordr). 2024;47:1879-95
85. Sun L, Jiang G, Ng YY, Xiao L, Du Z, Wang S. et al. T cells with split CARs specific for NKG2D ligands and PD-L1 exhibit improved selectivity towards monocyte-derived cells while effective in eliminating acute myeloid leukaemia in vivo. J Cancer Res Clin Oncol. 2023;149:10189-201
86. Perriello VM, Rotiroti MC, Pisani I, Galimberti S, Alberti G, Pianigiani G. et al. IL-3-zetakine combined with a CD33 costimulatory receptor as a dual CAR approach for safer and selective targeting of AML. Blood Adv. 2023;7:2855-71
87. Alberti G, Arsuffi C, Pievani A, Salerno D, Mantegazza F, Dazzi F. et al. Engineering tandem CD33xCD146 CAR CIK (cytokine-induced killer) cells to target the acute myeloid leukemia niche. Front Immunol. 2023;14:1192333
88. Jin X, Xie D, Sun R, Lu W, Xiao X, Yu Y. et al. CAR-T cells dual-target CD123 and NKG2DLs to eradicate AML cells and selectively target immunosuppressive cells. Oncoimmunology. 2023;12:2248826
89. Wang X, Dong Z, Awuah D, Chang WC, Cheng WA, Vyas V. et al. CD19/BAFF-R dual-targeted CAR T cells for the treatment of mixed antigen-negative variants of acute lymphoblastic leukemia. Leukemia. 2022;36:1015-24
90. Atilla E, Benabdellah K. The Black Hole: CAR T Cell Therapy in AML. Cancers (Basel). 2023;15:2713
91. Teppert K, Yonezawa Ogusuku IE, Brandes C, Herbel V, Winter N, Werchau N. et al. CAR'TCR-T cells co-expressing CD33-CAR and dNPM1-TCR as superior dual-targeting approach for AML treatment. Mol Ther Oncol. 2024;32:200797
92. Dao T, Xiong G, Mun SS, Meyerberg J, Korontsvit T, Xiang J. et al. A dual-receptor T-cell platform with Ab-TCR and costimulatory receptor achieves specificity and potency against AML. Blood. 2024;143:507-21
93. Li D, Qin J, Zhou T, Li Y, Cheng X, Chen Z. et al. Bispecific GPC3/PD-1 CAR-T cells for the treatment of HCC. Int J Oncol. 2023;62:53
94. Wang H, Wang X, Ye X, Ju Y, Cao N, Wang S. et al. Nonviral mcDNA-mediated bispecific CAR T cells kill tumor cells in an experimental mouse model of hepatocellular carcinoma. BMC Cancer. 2022;22:814
95. Li K, Qian S, Huang M, Chen M, Peng L, Liu J. et al. Development of GPC3 and EGFR-dual-targeting chimeric antigen receptor-T cells for adoptive T cell therapy. Am J Transl Res. 2021;13:156-67
96. Jiang W, Li T, Guo J, Wang J, Jia L, Shi X. et al. Bispecific c-Met/PD-L1 CAR-T Cells Have Enhanced Therapeutic Effects on Hepatocellular Carcinoma. Front Oncol. 2021;11:546586
97. Chen C, Li K, Jiang H, Song F, Gao H, Pan X. et al. Development of T cells carrying two complementary chimeric antigen receptors against glypican-3 and asialoglycoprotein receptor 1 for the treatment of hepatocellular carcinoma. Cancer Immunol Immunother. 2017;66:475-89
98. Mun SS, Meyerberg J, Peraro L, Korontsvit T, Gardner T, Malviya M. et al. Dual targeting ovarian cancer by Muc16 CAR T cells secreting a bispecific T cell engager antibody for an intracellular tumor antigen WT1. Cancer Immunol Immunother. 2023;72:3773-86
99. Li T, Wang J. Therapeutic effect of dual CAR-T targeting PDL1 and MUC16 antigens on ovarian cancer cells in mice. BMC Cancer. 2020;20:678
100. Jiang G, Ng YY, Tay JCK, Du Z, Xiao L, Wang S. et al. Dual CAR-T cells to treat cancers co-expressing NKG2D and PD1 ligands in xenograft models of peritoneal metastasis. Cancer Immunol Immunother. 2023;72:223-34
101. Shu R, Evtimov VJ, Hammett MV, Nguyen NN, Zhuang J, Hudson PJ. et al. Engineered CAR-T cells targeting TAG-72 and CD47 in ovarian cancer. Mol Ther Oncolytics. 2021;20:325-41
102. Hou AJ, Shih RM, Uy BR, Shafer A, Chang ZNL, Comin-Anduix B. et al. IL-13Rα2/TGF-β bispecific CAR-T cells counter TGF-β-mediated immune suppression and potentiate anti-tumor responses in glioblastoma. Neuro Oncol. 2024;26:1850-66
103. Saleh HA, Mitwasi N, Ullrich M, Kubeil M, Toussaint M, Deuther-Conrad W. et al. Specific and safe targeting of glioblastoma using switchable and logic-gated RevCAR T cells. Front Immunol. 2023;14:1166169
104. Ren Y, Bao X, Feng M, Xing B, Lian W, Yao Y. et al. CD87-targeted BiTE and CAR-T cells potently inhibit invasive nonfunctional pituitary adenomas. Sci China Life Sci. 2024;67:2169-85
105. Tapia-Galisteo A, Sánchez-Rodríguez I, Narbona J, Iglesias-Hernández P, Aragón-García S, Jiménez-Reinoso A. et al. Combination of T cell-redirecting strategies with a bispecific antibody blocking TGF-β and PD-L1 enhances antitumor responses. Oncoimmunology. 2024;13:2338558
106. Timpanaro A, Piccand C, Dzhumashev D, Anton-Joseph S, Robbi A, Moser J. et al. CD276-CAR T cells and Dual-CAR T cells targeting CD276/FGFR4 promote rhabdomyosarcoma clearance in orthotopic mouse models. J Exp Clin Cancer Res. 2023;42:293
107. Liu Y, Zheng Y, Deng T, Huang Y, Liu Z, Zhan B. et al. Oncolytic herpes simplex virus delivery of dual CAR targets of CD19 and BCMA as well as immunomodulators to enhance therapeutic efficacy in solid tumors combined with CAR T cell therapy. Front Oncol. 2022;12:1037934
108. Yuan X, Sun Z, Yuan Q, Hou W, Liang Q, Wang Y. et al. Dual-function chimeric antigen receptor T cells targeting c-Met and PD-1 exhibit potent anti-tumor efficacy in solid tumors. Invest New Drugs. 2021;39:34-51
109. Zhang E, Yang P, Gu J, Wu H, Chi X, Liu C. et al. Recombination of a dual-CAR-modified T lymphocyte to accurately eliminate pancreatic malignancy. J Hematol Oncol. 2018;11:102
110. Testa U, Pelosi E, Castelli G. Clinical value of identifying genes that inhibit hepatocellular carcinomas. Expert Rev Mol Diagn. 2022;22:1009-35
111. Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391:1301-14
112. Lai JP, Sandhu DS, Yu C, Han T, Moser CD, Jackson KK. et al. Sulfatase 2 up-regulates glypican 3, promotes fibroblast growth factor signaling, and decreases survival in hepatocellular carcinoma. Hepatology. 2008;47:1211-22
113. Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol. 2018;15:599-616
114. Li L, Wang H. Heterogeneity of liver cancer and personalized therapy. Cancer Lett. 2016;379:191-7
115. Yamauchi N, Watanabe A, Hishinuma M, Ohashi K, Midorikawa Y, Morishita Y. et al. The glypican 3 oncofetal protein is a promising diagnostic marker for hepatocellular carcinoma. Mod Pathol. 2005;18:1591-8
116. Baumhoer D, Tornillo L, Stadlmann S, Roncalli M, Diamantis EK, Terracciano LM. Glypican 3 expression in human nonneoplastic, preneoplastic, and neoplastic tissues: a tissue microarray analysis of 4,387 tissue samples. Am J Clin Pathol. 2008;129:899-906
117. Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W. et al. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016;76:4124-35
118. Zou B, Liu X, Zhang B, Gong Y, Cai C, Li P. et al. The Expression of FAP in Hepatocellular Carcinoma Cells is Induced by Hypoxia and Correlates with Poor Clinical Outcomes. J Cancer. 2018;9:3278-86
119. Fitzgerald AA, Weiner LM. The role of fibroblast activation protein in health and malignancy. Cancer Metastasis Rev. 2020;39:783-803
120. Sun L, Gao F, Gao Z, Ao L, Li N, Ma S. et al. Shed antigen-induced blocking effect on CAR-T cells targeting Glypican-3 in Hepatocellular Carcinoma. J Immunother Cancer. 2021;9:e001875
121. Wu M, Huang Q, Xie Y, Wu X, Ma H, Zhang Y. et al. Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation. J Hematol Oncol. 2022;15:24
122. Shen N, Yang C, Zhang X, Tang Z, Chen X. Cisplatin nanoparticles possess stronger anti-tumor synergy with PD1/PD-L1 inhibitors than the parental drug. Acta Biomater. 2021;135:543-55
123. Daveau M, Scotte M, François A, Coulouarn C, Ros G, Tallet Y. et al. Hepatocyte growth factor, transforming growth factor alpha, and their receptors as combined markers of prognosis in hepatocellular carcinoma. Mol Carcinog. 2003;36:130-41
124. Ito Y, Takeda T, Sakon M, Tsujimoto M, Higashiyama S, Noda K. et al. Expression and clinical significance of erb-B receptor family in hepatocellular carcinoma. Br J Cancer. 2001;84:1377-83
125. Kroeger PT Jr, Drapkin R. Pathogenesis and heterogeneity of ovarian cancer. Curr Opin Obstet Gynecol. 2017;29:26-34
126. Galon J, Bruni D. Tumor Immunology and Tumor Evolution: Intertwined Histories. Immunity. 2020;52:55-81
127. Egen JG, Ouyang W, Wu LC. Human Anti-tumor Immunity: Insights from Immunotherapy Clinical Trials. Immunity. 2020;52:36-54
128. Sundar S, Neal RD, Kehoe S. Diagnosis of ovarian cancer. Bmj. 2015;351:h4443
129. Stanczak MA, Siddiqui SS, Trefny MP, Thommen DS, Boligan KF, von Gunten S. et al. Self-associated molecular patterns mediate cancer immune evasion by engaging Siglecs on T cells. J Clin Invest. 2018;128:4912-23
130. Belisle JA, Horibata S, Jennifer GA, Petrie S, Kapur A, André S. et al. Identification of Siglec-9 as the receptor for MUC16 on human NK cells, B cells, and monocytes. Mol Cancer. 2010;9:118
131. Oka Y, Tsuboi A, Kawakami M, Elisseeva OA, Nakajima H, Udaka K. et al. Development of WT1 peptide cancer vaccine against hematopoietic malignancies and solid cancers. Curr Med Chem. 2006;13:2345-52
132. Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225-74
133. Conejo-Garcia JR, Benencia F, Courreges MC, Khang E, Zhang L, Mohamed-Hadley A. et al. Letal, A tumor-associated NKG2D immunoreceptor ligand, induces activation and expansion of effector immune cells. Cancer Biol Ther. 2003;2:446-51
134. Conejo-Garcia JR, Benencia F, Courreges MC, Gimotty PA, Khang E, Buckanovich RJ. et al. Ovarian carcinoma expresses the NKG2D ligand Letal and promotes the survival and expansion of CD28- antitumor T cells. Cancer Res. 2004;64:2175-82
135. Yuan B, Wang G, Tang X, Tong A, Zhou L. Immunotherapy of glioblastoma: Recent advances and future prospects. Hum Vaccin Immunother. 2022;18:2055417
136. Vik-Mo EO, Nyakas M, Mikkelsen BV, Moe MC, Due-Tønnesen P, Suso EM. et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother. 2013;62:1499-509
137. Agosti E, Zeppieri M, De Maria L, Tedeschi C, Fontanella MM, Panciani PP. et al. Glioblastoma Immunotherapy: A Systematic Review of the Present Strategies and Prospects for Advancements. Int J Mol Sci. 2023;24:15037
138. Brown CE, Warden CD, Starr R, Deng X, Badie B, Yuan YC. et al. Glioma IL13Rα2 is associated with mesenchymal signature gene expression and poor patient prognosis. PLoS One. 2013;8:e77769
139. Saikali S, Avril T, Collet B, Hamlat A, Bansard JY, Drenou B. et al. Expression of nine tumour antigens in a series of human glioblastoma multiforme: interest of EGFRvIII, IL-13Ralpha2, gp100 and TRP-2 for immunotherapy. J Neurooncol. 2007;81:139-48
140. Newman JP, Wang GY, Arima K, Guan SP, Waters MR, Cavenee WK. et al. Interleukin-13 receptor alpha 2 cooperates with EGFRvIII signaling to promote glioblastoma multiforme. Nat Commun. 2017;8:1913
141. Ikushima H, Todo T, Ino Y, Takahashi M, Miyazawa K, Miyazono K. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell. 2009;5:504-14
142. Bruna A, Darken RS, Rojo F, Ocaña A, Peñuelas S, Arias A. et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell. 2007;11:147-60
143. Lan Y, Yeung TL, Huang H, Wegener AA, Saha S, Toister-Achituv M. et al. Colocalized targeting of TGF-β and PD-L1 by bintrafusp alfa elicits distinct antitumor responses. J Immunother Cancer. 2022;10:e004122
144. Lind H, Gameiro SR, Jochems C, Donahue RN, Strauss J, Gulley JM. et al. Dual targeting of TGF-β and PD-L1 via a bifunctional anti-PD-L1/TGF-βRII agent: status of preclinical and clinical advances. J Immunother Cancer. 2020;8:e000433
145. Luke JJ, Patel MR, Blumenschein GR, Hamilton E, Chmielowski B, Ulahannan SV. et al. The PD-1- and LAG-3-targeting bispecific molecule tebotelimab in solid tumors and hematologic cancers: a phase 1 trial. Nat Med. 2023;29:2814-24
146. Zou R, Zhou X, Liu H, Wang P, Xia F, Kang L. et al. Long-term Complete Remission of Decitabine-Primed Tandem CD19/CD22 CAR-T Therapy with PD-1 and BTK Inhibitors Maintenance in a Refractory Primary Central Nervous System Lymphoma Patient. Cancer Res Treat. 2023;55:1363-8
147. Ruella M, Klichinsky M, Kenderian SS, Shestova O, Ziober A, Kraft DO. et al. Overcoming the Immunosuppressive Tumor Microenvironment of Hodgkin Lymphoma Using Chimeric Antigen Receptor T Cells. Cancer Discov. 2017;7:1154-67
148. Senior K. Making CAR T-cell therapies more affordable. Lancet. 2025;405:187-8
149. Tong C, Zhang Y, Liu Y, Ji X, Zhang W, Guo Y. et al. Optimized tandem CD19/CD20 CAR-engineered T cells in refractory/relapsed B-cell lymphoma. Blood. 2020;136:1632-44
150. Qu C, Zou R, Wang P, Zhu Q, Kang L, Ping N. et al. Decitabine-primed tandem CD19/CD22 CAR-T therapy in relapsed/refractory diffuse large B-cell lymphoma patients. Front Immunol. 2022;13:969660
151. Mulvey A, Trueb L, Coukos G. Novel strategies to manage CAR-T cell toxicity. Nat Rev Drug Discov. 2025
152. Freyer CW, Porter DL. Cytokine release syndrome and neurotoxicity following CAR T-cell therapy for hematologic malignancies. J Allergy Clin Immunol. 2020;146:940-8
153. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C. et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673-83
154. Lu LL, Xiao SX, Lin ZY, Bai JJ, Li W, Song ZQ. et al. GPC3-IL7-CCL19-CAR-T primes immune microenvironment reconstitution for hepatocellular carcinoma therapy. Cell Biol Toxicol. 2023;39:3101-19
155. Tousley AM, Rotiroti MC, Labanieh L, Rysavy LW, Kim WJ. Co-opting signalling molecules enables logic-gated control of CAR T cells. Nature. 2023;615:507-16
156. Dimitri A, Herbst F, Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer. 2022;21:78
157. Albarrán-Fernández V, Angelats L, Delgado J, Gros A. Unlocking the potential of engineered immune cell therapy for solid tumors. Nat Commun. 2025;16:1144
Author contact
![]() Corresponding authors: Jinbiao Liu, No. 28 Nanli Road, Hubei Key Laboratory of Industrial Microbiology, School of Life and Health Sciences, Hubei University of Technology, Wuhan, Hubei, 430068, China (Email: jinbiaoliuedu.cn), Yao Xu, No. 2 Huangjiahu West Road, Institute of Biology and Medicine, College of Life Science and Health, Wuhan University of Science and Technology, Wuhan, Hubei, 430081, China (Email: xuyao0307edu.cn; Tel.: +86-02768897343).
Corresponding authors: Jinbiao Liu, No. 28 Nanli Road, Hubei Key Laboratory of Industrial Microbiology, School of Life and Health Sciences, Hubei University of Technology, Wuhan, Hubei, 430068, China (Email: jinbiaoliuedu.cn), Yao Xu, No. 2 Huangjiahu West Road, Institute of Biology and Medicine, College of Life Science and Health, Wuhan University of Science and Technology, Wuhan, Hubei, 430081, China (Email: xuyao0307edu.cn; Tel.: +86-02768897343).