Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusions
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(7):947-959. doi:10.7150/ijbs.7.947 This issue Cite
Research Paper
Molecular Characterization of Transcriptome-wide Interactions between Highly Pathogenic Porcine Reproductive and Respiratory Syndrome Virus and Porcine Alveolar Macrophages in vivo
Ping Zhou1, Shanli Zhai1, Xiang Zhou1, Ping Lin1, Tengfei Jiang1, Xueying Hu2, Yunbo Jiang3, Bin Wu3, Qingde Zhang1, Xuewen Xu1, Jin-ping Li4, Bang Liu1, ![]()
1. Key Lab of Agricultural Animal Genetics, Breeding, and Reproduction of Ministry of Education & Key Lab of Swine Genetics and Breeding of Ministry of Agriculture, Huazhong Agricultural University, Wuhan 430070, Hubei, China;
2. Pathology Lab of Animal, College of Veterinary Medicine, Huazhong Agricultural University, Wuhan 430070, Hubei, China;
3. State Key Laboratory of Agricultural Microbiology, College of Veterinary Medicine, Huazhong Agricultural University, Wuhan 430070, Hubei, China;
4. Department of Medical Biochemistry and Microbiology, Biomedical Center, Uppsala University, Box 582, SE-751 23 Uppsala, Sweden.
Received 2011-6-5; Accepted 2011-7-30; Published 2011-8-7
Abstract
Porcine reproductive and respiratory syndrome virus (PRRSV) infects mainly the porcine alveolar macrophages (PAMs) and causes porcine reproductive and respiratory syndrome (PRRS). Previous studies have analyzed the global gene expression profiles of lung tissue in vivo and PAMs in vitro following infection with PRRSV, however, transcriptome-wide understanding of the interaction between highly pathogenic PRRSV (HP-PRRSV) and PAMs in vivo has not yet been established. In this study, we employed Affymetrix microarrays to investigate the gene expression patterns of PAMs isolated from Tongcheng piglets (a Chinese indigenous breed) after infection with HP-PRRSV. During the infection, Tongcheng piglets exhibited typical clinical signs, e.g. fever, asthma, coughing, anorexia, lethargy and convulsion, but displayed mild regional lung damage at 5 and 7 dpi. Microarray analysis revealed that HP-PRRSV infection has affected PAMs in expression of the important genes involved in cytoskeleton and exocytosis organization, protein degradation and folding, intracellular calcium and zinc homeostasis. Several potential antiviral strategies might be employed in PAMs, including upregulating IFN-induced genes and increasing intracellular zinc ion concentration. And inhibition of the complement system likely attenuated the lung damage during HP-PRRSV infection. Transcriptomic analysis of PAMs in vivo could lead to a better understanding of the HP-PRRSV-host interaction, and to the identification of novel antiviral therapies and genetic components of swine tolerance/susceptibility to HP-PRRS.
Keywords: microarray, HP-PRRSV-host interaction, infection, pulmonary alveolar macrophage, antiviral strategy
Introduction
Porcine reproductive and respiratory syndrome (PRRS), caused by PRRS virus (PRRSV) which belongs to the genus Arterivirus of the family Arteriviridae, is the most economically significant disease effecting commercially bred pigs world-wide [1]. This disease is characterized by anorexia, increased late-term abortions, increased number of stillborn pigs, mummified fetuses, weak live-born piglets, increased pre-weaning mortality, and delayed return to estrus [2]. In vivo, PRRSV productive infection occurs predominately in alveolar macrophages of the lung [3], followed by viremia and subsequent interstitial pneumonia within 3 days [4]. It was hypothesized that respiratory pathology, especially lung damage during PRRSV infection, results from an overproduction of pro-inflammatory cytokines in the lungs [5].
Genome-wide transcriptional responses of lungs of Landrace×Yorkshire crossbred piglets to a classical North American type PRRSV strain infection was analyzed by Solexa/Illumina's Digital Gene Expression (DGE) System, which is a tag-based high-throughput transcriptome sequencing method [6]. This systematic analysis of the pulmonary gene expression profiles suggested that upregulation expression of pro-inflammatory cytokines, chemokines, adhesion molecules and inflammatory enzymes and inflammatory cells, antibodies, complement activation were likely to result in the development of inflammatory responses during PRRSV infection processes [6]. Another high-throughput deep sequencing was performed focusing on the pulmonary gene expression profiles after a highly pathogenic-PRRSV (HP-PRRSV) strain infection [7]. The system analysis of the pulmonary gene expression provides a comprehensive basis for better understanding the pathogenesis of HP-PRRSV [7]. Because PRRSV infection occurs predominately in porcine alveolar macrophages (PAMs) [3], the interaction between PRRSV and PAMs have been studied systematically by high-throughput research methods in vitro. PAMs, lavaged from six piglets, were challenged with the Lelystad PRRSV strain in vitro, and the gene expression of the PAMs was investigated using Affymetrix microarrays [8]. The result suggested that the expression of beta interferon 1 (IFN-β), but not of IFN-α, was strongly upregulated in the early stage of PRRSV infection [8]. Besides microarray, Serial Analysis of Gene Expression (SAGE) was also employed to examine the global expression of genes in PRRSV-infected PAMs in vitro [9]. These studies have provided global gene expression profiles of lung tissue in vivo and PAMs in vitro following infection with PRRSV; however, transcriptome-wide understanding of the interaction between PRRSV and PAMs in vivo has not yet been established.
In 2006, an unparalleled large-scale outbreak of highly pathogenic PRRS (HP-PRRS) occured in many areas of China. This outbreak affected more than 2 millions pigs and produced approximately 0.4 million fatal cases [10]. In this study, laboratory infection was performed in Tongcheng piglets (a Chinese indigenous breed living in Tongcheng county of Hubei province) using PRRSV stain WUH3 [11], a highly pathogenic PRRSV isolated in China during the pandemic period of HP-PRRS in 2006. We also employed Affymetrix microarrays to investigate the gene expression patterns of PAMs isolated from the piglets after infection. The current study aims at better understanding the interaction between HP-PRRSV and the host PAMs, which may lead to the identification of key host factors for tolerance/susceptibility to the virus and the finding of novel targets for antiviral therapies.
Materials and methods
Animals and experimental design
All animal procedures were performed according to protocols approved by the Biological Studies Animal Care and Use Committee of Hubei Province, China. Piglets used in this study were free from PRRSV, pseudorabies virus (PRV) and porcine circovirus type 2 (PCV2) determined by ELISA test for serum antibodies. Twelve 5-week-old Tongcheng boars (a Chinese indigenous breed) were obtained from three litters (four piglets per litter), and raised in pathogen-free facilities. To perform a paired experiment, individuals within a full-sib litter were separated into two groups: one infected group and one control group with 6 piglets in each group. The infected groups were challenged with PRRSV-WUH3 (3 ml/15 kg, 10-5 TCID50/ml) by intramuscular inoculation. Slaughters were carried out at 0 day post-infection (dpi) for uninfected (control) groups, and at 5 or 7 dpi for infected groups. Rectal temperature and clinical signs were recorded daily during the experiment. The serum samples for viremia detection were collected daily from all animals (one ml blood per sampling point). The PAMs for microarray analysis were collected by bronchoalveolar lavage from three uninfected pigs and three infected pigs at 5 dpi.
Gross and microscopic lung lesions
Post-mortem examinations were performed on all pigs. Macroscopic lung lesions were given a subjective score to estimate the percentage of the lung affected by pneumonia, following a scoring system described previously [12, 13]. For histopathology analysis, samples of the apical segment of the lower lung lobes were collected and fixed in 4% paraformalclehyde for 24 h. Fixed samples were dehydrated, embedded in paraffin, sectioned into 4 μm and stained with hematoxylin and eosin. Sections were examined by light microscopy.
Viral load determination
For viremia detection, serum samples were collected daily from all pigs. Total viral RNA was extracted from 200 μl serum using TRIzol Reagent (Invitrogen, Carlsbad, CA). cDNA was synthesized using oligo(dT)15 primer, M-MLV Reverse Transcriptase (Promega, Madison, WI) in 50 μl reaction mixture according to the manufacturer's instructions. Absolute quantitative-PCR (Q-PCR) was performed using primers specific to the ORF7 of PRRSV (sense: 5'-TCA GCT GTG CCA AAT GCT GG-3'; antisense: 5'-AAA TGG GGC TTC TCC GGG TTT T-3'). For absolute quantification, the pET-18M plasmid of the known copy number containing the ORF7 fragment generated standard curve. Viral copies per ml of the unknown samples were determined by linear extrapolation of the Ct value plotted against the standard curve [14].
RNA extraction and microarray hybridization
TRIzol (Invitrogen) was used for RNA extractions following the manufacturer's instructions. RNA integrity and concentration were evaluated by denaturing formaldehyde gel electrophoresis and Agilent 2100 Bioanalyzer. The RNA samples were sent to GeneTech Biotechnology Limited Company (Shanghai, China) for hybridization to the porcine Affymetrix GeneChip (Affymetrix, Santa Clara, CA). A total of 6 microarray analyses were conducted using the procedure described previously [15].
Microarray data analysis
The raw data (Affymetrix GeneChip Scanner 3000) was converted to gene signal files by MAS 5.0 (Microarray Suite Version 5.0, Affymetrix). The data points were normalized between slides using the quantile normalization method used by Bolstad et al. [16]. The differentially expressed genes were selected using the SAM (Significance Analysis of Microarrays) package (http://www-stat.stanford.edu/~tibs/SAM/), and the false discovery rate (FDR) values were generated using permutations of the repeated measurements to estimate the percentage of genes identified by chance. In the experiment, SAM settings were adjusted for a two class paired analysis, using one hundred permutations to calculate the differentially expressed gene list. The fold-change of 1.5 and a false discovery rate of approximately 5% were set as a threshold. All data are MIAME compliant and have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE22782 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE22782). Differential gene expressions were performed for hierarchical cluster (Ver.3.0) and TreeView (Ver.1.60) analyses [17]. The functional annotation of differentially expressed genes was performed by the DAVID (The Database for Annotation, Visualization and Integrated Discovery) gene annotation tool (http://david.abcc.ncifcrf.gov/) [18], as well as by referring to a previous work [19].
Q-PCR
The RNA samples prepared for microarray analysis were also used for Q-PCR verification. Reverse transcriptions were performed using M-MLV Reverse Transcriptase (Promega) according to the manufacturer's instructions. The primers were designed with the Primer Premier 5.0 program. The RPL32 gene was used as the internal control [20]. The primer sequences, melting temperatures and product sizes are shown in Table 1. Q-PCR was performed on the LightCycler 480Ⅱ (Roche, Basel, Sweden) using SYBR Green Realtime PCR Master Mix (TOYOBO CO., LTD, Japan) as the readout. Data was analyzed by the 2-ΔΔCT method [21]. The data analysis procedure was performed as described previously [15].
Primers used for Q-PCR validation
| Gene | Primer sequence (5'-3') | Target size (bp) | Tm (℃)a |
|---|---|---|---|
| C3 | Forward: AAACTAAAGGAGGGGGGACACT Reverse: CTTGGCATACATCACCATCAGG | 133 | 60 |
| CCL2 | Forward: AACTTGCCCTAAATACCCTCAGA Reverse: GGAAAGCAATGTGCCCAAGTC | 179 | 61 |
| DDIT3 | Forward: ACGGCTCAAGCAGGAAATC Reverse: CACTGGTAAGAAGGTGGTTGGT | 173 | 58 |
| EMP1 | Forward: CTCAGATGCGGGACAAGGA Reverse: AAGACCACGAGGGAGACGA | 156 | 58 |
| GLRX2 | Forward: TACGGAAGCCAGTTTCAAGAC Reverse: CTTGGTGAAGCCTATGAGTGTC | 118 | 58 |
| TNF | Forward: CATCGCCGTCTCCTACCA Reverse: CCCAGATTCAGCAAAGTCCA | 199 | 58 |
| RPL32b | Forward: CGGAAGTTTCTGGTACACAATGTAA Reverse: TGGAAGAGACGTTGTGAGCAA | 94 | 58-61 |
aThe annealing temperature represents the optimal temperature during quantitative PCR;
bRNA levels of RPL32 was assayed for normalization during quantitative PCR.
Results
Clinical and pathological features of HP-PRRSV-infected piglets
After infection with PRRSV-WUH3, the piglets presented typical clinical signs, e.g. fever, asthma, coughing, anorexia, lethargy and convulsion. The average rectal temperature rose to above 40.5 ºC at 2 dpi and seemed to peak at 5 dpi. The two piglets surviving at 5 dpi showed a slight decrease of rectal temperature in the following two days (Figure 1A). To assess the replication and spread of HP-PRRSV, the viral copy number/ml in serum was determined by absolute real-time quantitative-PCR (Figure 1B). The level of viremia increased rapidly during the first two days post-infection, then increased slowly from 3 to 5 dpi, and approached the plateau phase at 6 or 7 dpi.
Laboratory infection of Tongcheng piglets with HP-PRRSV. A: Rectal temperature of the piglets; B: Viral copy numbers per ml serum determined by absolute real-time quantitative PCR; C: Lung morphology of piglets slaughtered at 0, 5 or 7 dpi; D: Gross lung lesion scores of the piglets. The scores of 0 to 100 denote the different severity of gross lung lesion, from intact to totally damaged; E and F: Paraffin sections of the apex of lung at 0 dpi (E) and 5 dpi (F), stained with hematoxylin and eosin. Scale bars indicate 50 μm.
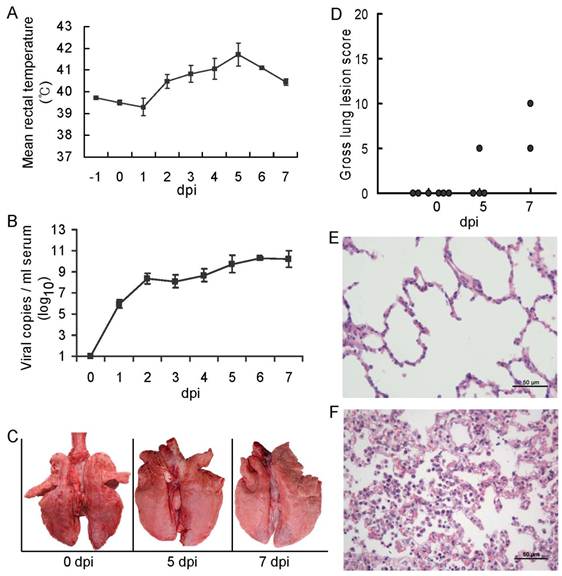
Pathologic examination was carried out on the animals. Macroscopic examination detected a mild lung lesion at the apical segment of the lower lobes at 5 and 7 dpi (Figure 1C). For estimating the severity of the pneumonia, gross lung lesion scores were made based on the method described previously [12, 13]. The low scores indicated a mild regional lung damage at 5 and 7 days after HP-PRRSV infection (Figure 1D). As compared with the uninfected group (Figure 1E), microscopic examination detected a certain extent of congestion as well as interstitial infiltration of leukocytes in the lungs of infected piglets (Figure 1F).
Transcriptome analysis of the virus-host cell interaction in vivo
PAMs samples collected from three infected piglets at 5 dpi and three uninfected piglets were analyzed. A total of 12,775 transcripts (53% of all probesets) were expressed in infected and non-infected PAMs (Supplementary Table 1). After quantile normalization, 321 genes were identified as differentially expressed (DE) genes, with 219 being upregulated and 102 being downregulated, under the threshold of fold change (FC) of 1.5 or greater and a false discovery rate (FDR) of approximately 5% (Figure 2A and Supplementary Table 2).
Microarray analysis of PAMs' transcriptional responses to HP-PRRSV infection. A: Hierarchical clustering analysis of gene expression profiles pre- and post-infection. Each column represents one piglet, and each horizontal line refers to a gene. Color legend is on the top-left of the figure. Red indicates genes with a greater expression relative to the geometrical means, green indicates genes with a lower expression relative to the geometrical means; B: Biological process Gene Ontology (GO) analysis of 166 differentially expressed genes. Many categories shared the same transcripts.
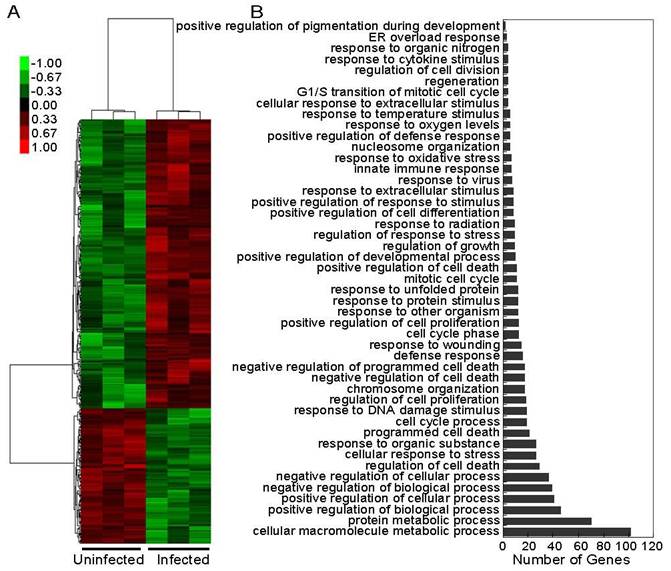
Based on the Database for Annotation, Visualization and Integrated Discovery (DAVID), 166 of the DE genes were classified into 47 categories, many of which shared the same genes, according to their functional correlation (Figure 2B and Supplementary Table 3). The majority of the genes related to the virus-host cell interaction could be assigned into the categories including cell death and apoptosis related, response to wounding, response to unfolded protein, response to oxidative stress, response to virus, innate immune response, response to cytokine stimulus, and endoplasmic reticulum (ER) overload response. Other DE genes that were not classified by DAVID were taken into account for further analysis below.
Sensing the HP-PRRSV infection by PAMs
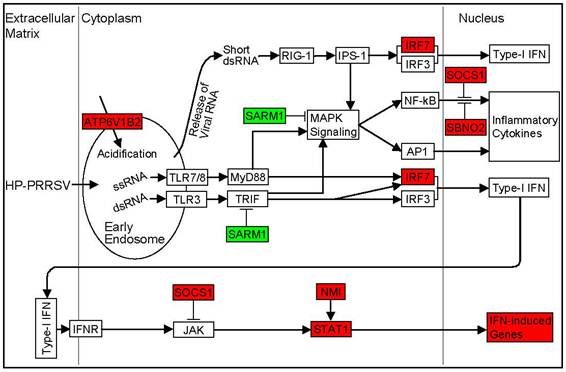
In macrophages, PRRSV entry into the host cell is mediated by heparan sulphate proteoglycans and the receptor sialoadhesin. Upon a pH drop, PRRSV is uncoated and its genome is released from the endosomes into the cytoplasm, which allows virus replication [22]. After HP-PRRSV infection the ATP6V1B2 gene, which encodes a component of vacuolar ATPase (V-ATPase) that mediates acidification of endosomal organelles [23], was upregulated (Figure 3). SARM1, a negative regulator of TRIF-dependent Toll-like receptor (TLR) signaling [24] and MAPK phosphorylation [25], was significantly downregulated (Figure 3). SBNO2, a potent inhibitor of NF-κB [26], and SOCS1 which limits NF-κB signaling by decreasing p65 stability within the cell nucleus [27], were upregulated (Figure 3). Upon HP-PRRSV infection, IRF7 was found to be upregulated in PAMs at 5 dpi (Figure 3), however, no type-I IFN or IFN-γ induction was observed. A number of IFN-induced genes (IFI6, IFI16, IFIH1, IFIT2, IFIT3, IFITM3, GBP1, GBP2, MX1, GZMB, GZMH, ISG15, USP18, RSAD2, NMI) were upregulated (Table 2). JAK-STAT pathway seemed to be positively (STAT1 and NMI) as well as negatively (SOCS1) regulated during HP-PRRSV infection (Figure 3).
Interferon induced genes
| Gene symbol | Gene description | Affymetrix probe set ID | Fold Change |
|---|---|---|---|
| GBP1 | guanylate binding protein 1, interferon-inducible, 67kDa | Ssc.29054.1.A1_at | 3.51 |
| GBP2 | guanylate binding protein 2, interferon-inducible | Ssc.883.1.S1_a_at | 4.32 |
| GZMB | granzyme B (granzyme 2, cytotoxic T-lymphocyte-associated serine esterase 1) | Ssc.4939.2.S1_at | 19.07 |
| GZMH | granzyme H (cathepsin G-like 2, protein h-CCPX) | Ssc.25976.1.S1_at | 4.75 |
| IFI16 | interferon, gamma-inducible protein 16 | Ssc.10884.1.A1_at | 3.41 |
| IFI6 | Interferon, alpha-inducible protein 6 | Ssc.20101.1.S1_at | 4.34 |
| IFIH1 | interferon induced with helicase C domain 1 | Ssc.17894.1.A1_at | 3.53 |
| IFIT2 | interferon-induced protein with tetratricopeptide repeats 2 | Ssc.22620.1.S1_at | 6.16 |
| IFIT3 | interferon-induced protein with tetratricopeptide repeats 3 | Ssc.31140.1.S1_at | 4.17 |
| IFITM3 | interferon induced transmembrane protein 3 (1-8U) | Ssc.30956.1.A1_s_at | 6.05 |
| ISG15 | ISG15 ubiquitin-like modifier | Ssc.11557.1.A1_at | 7.89 |
| MX1 | myxovirus (influenza virus) resistance 1, interferon-inducible protein p78 (mouse) | Ssc.221.1.S1_at | 4.45 |
| NMI | N-myc (and STAT) interactor | Ssc.9572.1.A1_at | 1.81 |
| RSAD2 | radical S-adenosyl methionine domain containing 2 | Ssc.286.1.S1_s_at | 9.74 |
| USP18 | ubiquitin specific peptidase 18 | Ssc.336.1.S1_at | 3.1 |
Sensing the HP-PRRSV infection by host cell. PRRSV enters early endosomes but does not continue through the endocytic pathway to late endosomes [48]. The ATP6V1B2 gene, which encodes a component of vacuolar ATPase (V-ATPase) that mediates acidification of endosomal organelles [23], facilitates the uncoating of the virus. Viral nucleic acids could be sensed by Toll-like receptors (TLRs) pathway or RIG-I pathway both of which lead to type-I IFN induction by activating IRF3 and IRF7, and to inflammatory cytokines expression by activating the MAPK signaling pathway. Several IFN-induced genes were upregulated during HP-PRRSV infection, even though no induction of type-I IFN was observed. Red background in the gene box indicates upregulation of the gene expression, green indicates downregulation, and white indicates no change of the gene expression. Fold changes of the differentially expressed genes are 1.94 (ATP6V1B2), 0.31 (SARM1), 4.58 (IRF7), 1.79 (SOCS1), 1.59 (SBNO2), 1.81 (NMI), and 2.48 (STAT1), respectively.
Cytoskeleton and exocytosis organization in PAMs
Nine genes (S100A6, MARCKS, CACYBP, CCT6A, ARHE, CCT3, PTPN4, CCT7, and TWF1) related to actin and tubulin cytoskeleton organization were upregulated and three (RASSF8, ELMO1, and KIF11) were downregulated (Table 3). In addition, several exocytosis related genes (RSAD2, GSK3B, LMAN2L, EXOC2, SELS, COPZ2, SEC31L2, and SEC8L1) were differentially expressed in PAMs after HP-PRRSV infection (Table 3). RSAD2, encoding an IFN-induced protein which inhibits influenza A virus release from the plasma membrane of infected cells by affecting the formation of lipid rafts [28], was upregulated significantly. Vesicle trafficking between the Golgi apparatus and ER seemed to be restricted, because COPZ2, a member of the COPI coat which helps vesicles transport proteins from the cis end of the Golgi complex back to the rough ER [29], and SEC31L2, a component of the COPII vesicle coat that mediates vesicular traffic from the rough ER to the Golgi apparatus [30], were both downregulated.
DE genes and their relation to different aspects of the host cell organization
| Gene symbol | Gene description | Affymetrix probe set ID | Fold Change |
|---|---|---|---|
| Cytoskeleton | |||
| S100A6 | S100 calcium binding protein A6 | Ssc.21109.1.S1_a_at | 4.87 |
| MARCKS | MARCKS-like 1 | Ssc.1228.1.S1_at | 3.00 |
| CACYBP | calcyclin binding protein | Ssc.10299.1.A1_at | 2.28 |
| CCT6A | chaperonin containing TCP1, subunit 6A (zeta 1) | Ssc.11348.1.A2_at | 2.06 |
| ARHE | Rho family GTPase 3 | Ssc.4127.2.A1_at | 1.87 |
| CCT3 | chaperonin containing TCP1, subunit 3 (gamma | Ssc.902.1.S1_a_at | 1.71 |
| PTPN4 | protein tyrosine phosphatase, non-receptor type 4 (megakaryocyte) | Ssc.29697.1.A1_at | 1.70 |
| CCT7 | chaperonin containing TCP1, subunit 7 (gamma | Ssc.11002.1.S1_at | 1.64 |
| TWF1 | twinfilin, actin-binding protein, homolog 1 (Drosophila) | Ssc.7177.1.A1_at | 1.57 |
| RASSF8 | Ras association (RalGDS/AF-6) domain family (N-terminal) member | Ssc.15288.1.S1_at | 0.64 |
| ELMO1 | engulfment and cell motility 1 | Ssc.5648.1.A1_at | 0.63 |
| KIF11 | kinesin family member 11 | Ssc.30748.1.S1_at | 0.40 |
| Exocytosis and vesicle trafficking | |||
| RSAD2 | radical S-adenosyl methionine domain containing 2 | Ssc.286.1.S1_s_at | 9.74 |
| GSK3B | glycogen synthase kinase 3 beta | Ssc.30917.1.A1_at | 3.09 |
| LMAN2L | lectin, mannose-binding 2-like | Ssc.24211.1.S1_at | 1.71 |
| EXOC2 | exocyst complex component 2 | Ssc.7976.1.A1_at | 1.71 |
| SELS | selenoprotein S | Ssc.2739.1.S1_at | 1.71 |
| COPZ2 | coatomer protein complex, subunit zeta 2 | Ssc.4221.1.S1_at | 0.49 |
| SEC31L2 | SEC31 homolog B (S. cerevisiae) | Ssc.31182.1.A1_at | 0.43 |
| SEC8L1 | exocyst complex component 4 | Ssc.9822.1.A1_at | 0.25 |
| Ubiquitination and ISGylation | |||
| ISG15 | ISG15 ubiquitin-like modifier | Ssc.11557.1.A1_at | 7.89 |
| USP18 | ubiquitin specific peptidase 18 | Ssc.336.1.S1_at | 3.10 |
| CACYBP | calcyclin binding protein | Ssc.10299.1.A1_at | 2.28 |
| HERPUD1 | homocysteine-inducible, endoplasmic reticulum stress-inducible, ubiquitin-like domain member 1 | Ssc.12017.1.A1_at | 2.06 |
| CUL1 | cullin 1 | Ssc.30295.1.A1_at | 1.60 |
| HERC6 | hect domain and RLD 6 | Ssc.26483.1.A1_at | 1.66 |
| HERC3 | hect domain and RLD 3 | Ssc.9737.1.S1_at | 0.39 |
| G2E3 | G2/M-phase specific E3 ubiquitin protein ligase | Ssc.7317.1.A1_at | 0.58 |
| UBASH3B | ubiquitin associated and SH3 domain containing B | Ssc.25139.3.S1_at | 0.35 |
| Chaperones | |||
| NPM3 | nucleophosmin/nucleoplasmin 3 | Ssc.6196.1.S1_at | 2.23 |
| CCT3 | chaperonin containing TCP1, subunit 3 (gamma | Ssc.902.1.S1_a_at | 1.71 |
| CCT7 | chaperonin containing TCP1, subunit 7 (gamma | Ssc.11002.1.S1_at | 1.64 |
| CCT6A | chaperonin containing TCP1, subunit 6A (zeta 1) | Ssc.11348.1.A2_at | 2.06 |
| DNAJA1 | DnaJ (Hsp40) homolog, subfamily A, member 1 | Ssc.8473.1.S2_at | 2.03 |
| DNAJA4 | DnaJ (Hsp40) homolog, subfamily A, member 4 | Ssc.17243.1.S1_at | 11.31 |
| DNAJB1 | DnaJ (Hsp40) homolog, subfamily B, member 1 | Ssc.3502.1.S1_at | 3.46 |
| DNAJB2 | DnaJ (Hsp40) homolog, subfamily B, member 2 | Ssc.1180.1.S1_at | 1.54 |
| DNAJB4 | DnaJ (Hsp40) homolog, subfamily B, member 4 | Ssc.13345.1.A1_at | 8.78 |
| HSPA1B | heat shock 70kDa protein 1A | Ssc.5145.1.S1_at | 4.44 |
| HSPA4 | Heat shock 70kDa protein 4 | Ssc.3313.1.S1_at | 2.46 |
| HSPA4 | heat shock 70kDa protein 4 | Ssc.11197.1.S1_at | 1.98 |
| HSPA6 | heat shock 70kDa protein 6 (HSP70B') | Ssc.6728.1.S1_at | 1.60 |
| HSPD1 | heat shock 60kDa protein 1 (chaperonin) | Ssc.6719.1.A1_at | 1.83 |
| HSPH1 | heat shock 105kDa/110kDa protein 1 | Ssc.1231.1.A1_at | 2.94 |
| Intracellular calcium homeostasis | |||
| CKM | creatine kinase, muscle | Ssc.415.1.S1_at | 9.13 |
| KLB | klotho beta | Ssc.7252.1.A1_at | 2.77 |
| HERPUD1 | homocysteine-inducible, endoplasmic reticulum stress-inducible, ubiquitin-like domain member 1 | Ssc.12017.1.A1_at | 2.06 |
| TRPM4 | transient receptor potential cation channel, subfamily M, member 4 | Ssc.10022.1.A1_at | 1.81 |
| HAX1 | HCLS1 associated protein X-1 | Ssc.6188.1.S1_at | 1.79 |
| PIK3C2A | phosphoinositide-3-kinase, class 2, alpha polypeptide | Ssc.29061.1.A1_at | 0.58 |
| PIK3R2 | phosphoinositide-3-kinase, regulatory subunit 2 (beta) | Ssc.1677.1.A1_at | 0.47 |
| Zinc ion signaling | |||
| SLC39A14 | solute carrier family 39 (zinc transporter), member 14 | Ssc.19143.1.A1_at | 5.06 |
| ZDHHC9 | zinc finger, DHHC-type containing 9 | Ssc.16227.1.S1_at | 16.57 |
| ZFAND2A | zinc finger, AN1-type domain 2A | Ssc.5082.1.A1_at | 2.75 |
| ZCCHC6 | zinc finger, CCHC domain containing 6 | Ssc.21294.1.S1_at | 2.21 |
| ZCWPW1 | zinc finger, CW type with PWWP domain 1 | Ssc.31003.1.A1_at | 2.04 |
| ZFP2 | ZFP2 zinc finger protein 2 homolog | Ssc.7514.1.A1_at | 1.84 |
| ZNF258 | zinc finger, MYM-type 6 | Ssc.17765.1.S1_at | 0.50 |
Protein degradation and folding in PAMs
During HP-PRRSV infection, homeostasis of ISGylation, an ubiquitin-like modification, seemed to be re-established in PAMs by enhancing the expression of ISG15, an ubiquitin-like protein, and USP18, which is an ISG15 deconjugating protease (Table 3). Three E3 ubiquitin ligase genes (CACYBP, HERC6, CUL1) were upregulated, and two (HERC3, G2E3) were downregulated (Table 3). As expected, a large set of chaperone genes were upregulated, including heat shock 40 kDa protein (Hsp40) (DNAJA1, DNAJA4, DNAJB1, DNAJB2, and DNAJB4), Hsp60 (HSPD1), Hsp70 (HSPA1B, HSPA4, HSPA4, and HSPA6), Hsp105/110 (HSPH1), and subunits of chaperon in containing t-complex polypeptide 1 (CCT3, CCT7, and CCT6A), as well as NPM3, a molecular chaperone in the cell nucleus (Table 3).
Intracellular calcium and zinc homeostasis in PAMs
Upon HP-PRRSV infection, several DE genes were involved in the intracellular calcium homeostasis in PAMs (Table 3). After HP-PRRSV infection, zinc ion concentration in PAMs seemed to be increased, through upregulating the expression of SLC39A14 which encodes a zinc influx transporter [31] (Table 3). Several zinc finger protein encoding genes (ZDHHC9, ZFAND2A, ZCCHC6, ZCWPW1, ZFP2, ZNF258) were also identified as DE genes, and all of them were upregulated, except ZNF258 (Table 3).
Tissue remolding and inflammation
During HP-PRRSV infection, a set of DE genes involved in the dynamic regulation of the extracellular matrix and vascular permeability was identified (Table 4). Infiltration of leukocytes into pulmonary alveoli, as a sign of inflammation, was modulated by upregulating a small number of genes (CCL2, CCL4L, CCR5 and CSF1) (Table 4). Three genes, MPP1, PF4, and PPBP involved in neutrophil infiltration or activation [32-34], were all downregulated (Table 4). During HP-PRRSV infection, complement activation seemed to be inhibited, as expression of C3 and PFC, a positive regulator of complement activation, were downregulated, and CLU, encoding for a complement inhibitor, was upregulated (Table 4).
DE genes and their relation to tissue remolding and inflammation
| Gene symbol | Gene description | Affymetrix probe set ID | Fold Change |
|---|---|---|---|
| Extracellular matrix homeostasis | |||
| TIMP1 | TIMP metallopeptidase inhibitor 1 | Ssc.11784.1.S1_at | 4.48 |
| PLOD1 | procollagen-lysine 1, 2-oxoglutarate 5-dioxygenase 1 | Ssc.3530.1.A1_at | 3.74 |
| CSPG2 | versican | Ssc.5663.1.S1_at | 3.19 |
| ADAMTSL3 | ADAMTS-like 3 | Ssc.11976.1.A1_at | 2.72 |
| LAMC1 | laminin, gamma 1 (formerly LAMB2) | Ssc.1099.1.S1_at | 2.46 |
| ADAMTS20 | ADAM metallopeptidase with thrombospondin type 1 motif, 20 | Ssc.29491.1.A1_at | 2.40 |
| ST14 | suppression of tumorigenicity 14 (colon carcinoma) | Ssc.3285.1.S1_at | 1.84 |
| MMP28 | matrix metallopeptidase 28 | Ssc.29059.1.A1_at | 0.53 |
| NDST4 | N-deacetylase/N-sulfotransferase (heparan glucosaminyl) 4 | Ssc.13434.2.A1_at | 0.49 |
| TGFBI | transforming growth factor, beta-induced, 68kDa | Ssc.16671.1.S1_at | 0.36 |
| Vascular permeability | |||
| LRP11 | low density lipoprotein receptor-related protein 11 | Ssc.14282.1.A1_at | 4.37 |
| LRP8 | low density lipoprotein receptor-related protein 8, apolipoprotein e receptor | Ssc.31016.1.A1_at | 3.29 |
| SELL | selectin L | Ssc.22075.3.A1_at | 2.97 |
| SCPEP1 | serine carboxypeptidase 1 | Ssc.3037.3.A1_at | 2.21 |
| ANGPTL4 | angiopoietin-like 4 | Ssc.8980.1.A1_at | 0.51 |
| PLVAP | plasmalemma vesicle associated protein | Ssc.3645.1.S1_at | 0.19 |
| Complement system | |||
| CLU | clusterin | Ssc.11992.1.A1_at | 14.52 |
| PFC | complement factor properdin | Ssc.27474.1.S1_at | 0.49 |
| C3 | complement component 3 | Ssc.61.1.S1_at | 0.29 |
| Immune cells recruitment and activation | |||
| CCL2 | chemokine (C-C motif) ligand 2 | Ssc.657.1.A1_at | 14.17 |
| CSF1 | colony stimulating factor 1 (macrophage) | Ssc.6369.1.A1_at | 6.71 |
| CCL4L | chemokine (C-C motif) ligand 4-like 1 | Ssc.23797.1.S1_at | 4.86 |
| CLECL1 | C-type lectin-like 1 | Ssc.12825.1.A1_at | 2.66 |
| IL1RAP | interleukin 1 receptor accessory protein | Ssc.7864.1.A1_at | 2.49 |
| CCR5 | chemokine (C-C motif) receptor 5 | Ssc.26328.1.S1_at | 2.20 |
| MPP1 | membrane protein, palmitoylated 1, 55kDa | Ssc.19356.1.S1_at | 0.57 |
| PPBP | pro-platelet basic protein (chemokine (C-X-C motif) ligand 7) | Ssc.16228.1.S1_at | 0.39 |
| PF4 | platelet factor 4 (chemokine (C-X-C motif) ligand 4) | Ssc.24188.1.A1_at | 0.35 |
Confirmation of differential expression by Q-PCR
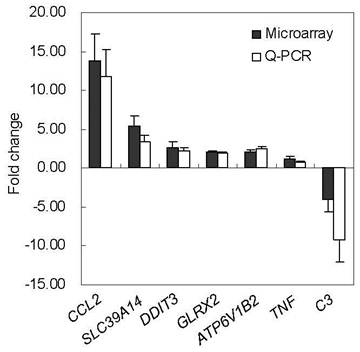
Seven genes (CCL2, SLC39A14, ATP6V1B2, C3, DDIT3, GLRX2 and TNF) were selected for Q-PCR assay to validate the changes in gene expression observed by microarray analysis. CCL2 was the main upregulated chemokine gene in this study (Table 4). Two upregulated genes, SLC39A14 and ATP6V1B2, were involved in intracellular zinc homeostasis and endosome acidification, respectively. The downregulated C3 gene is the core member of the complement system which seemed to be inhibited, according to our study (Table 4). The Q-PCR gene list also contained two DE genes (DDIT3, GLRX2) which were not referred to in the discussion, and TNF, an important cytokine gene, which was not differentially expressed in Tongcheng PAMs in response to HP-PRRSV infection. The changes of these genes, detected by microarray analysis, was in agreement with the Q-PCR validation (Figure 4).
Q-PCR validation of the microarray data. P values (T-test) of the Q-PCR data are 0.018 (CCL2), 0.039 (SLC39A14), 0.044 (DDIT3), 0.006 (GLRX2), 0.008 (ATP6V1B2), 0.508 (TNF) and 0.032 (C3), respectively. TNF is a non-differentially expressed gene.
Discussion
The results of this study showed that Tongcheng piglets exhibited typical clinical signs following infection with HP-PRRSV WUH3 strain. The lung damage caused by the infection was regional and mild at 5 and 7 dpi (Figure 1C and D), but further observation for a longer period of time was not performed in this study. The slow reproduction rate of the virus (viremia) at 3 to 7 dpi (Figure 1B) suggested a near balance between the viral replication and the defense mechanisms in the PAMs. Transcriptomic analysis of the PAMs at 5 dpi identified 321 DE genes under the filter of 1.5-fold change, and the number of upregulated genes (219) was greater than that of downregulated genes (102). In comparison, an in vitro transcriptomic analysis of PAMs revealed that only small numbers (no more than 100) of DE genes (threshold of 1.5-fold change) were identified at 1 to 12 hours post PRRSV infection, and the overall effect of PRRSV on the host transcription machinery was downegulation [8]. It is not sure whether there is a conversion of the overall effect of PRRSV on host transcription machinery from downregulation to upregulaton as time goes on, or the change is only the effect of the difference between in vitro and in vivo assays. As compared with the number of DE genes in this study, some thousands of DE genes (threshold of almost 1.5-fold change) were identified in lung tissues at 4 and 7 dpi following both PRRSV and HP-PRRSV infection, by high-throughput deep sequencing assays [6, 7]. This great number of DE genes might result from the huge amount of data obtained by the deep-sequencing method and from the many cell types in lung tissues.
PRRSV is considered to inhibit the synthesis of type-I IFNs and its signaling by blocking STAT1/STAT2 nuclear translocation [35]. However, it is also reported that PRRSV can phosphorylate IFN-regulatory factor 3 (IRF-3) and weakly activate the IFN-β promoter in MARC-145 cells in early infection, but the activations of IRF-3 and IFN-β promoter are rapidly inhibited in the following infection [36]. The induction of IFN-β mRNA, but not IFN-α mRNA, is observed in monocyte-derived dendritic cells and primary alveolar macrophages infected by PRRSV at 12 dpi [8, 37]. In some cases, even the expression of IFN-α can be detected in the lung [38] or serum [39] of pigs infected with PRRSV during the early days. Interestingly, in this study, no induction of type-I IFN was detected in PAMs at 5 dpi (Figure 3), whereas a series of IFN induced genes that are critical for the cell to defend itself against viral infection, were upregulated (Table 2). Similar results were shown in lung tissues at 4 and 7 dpi following both PRRSV and HP-PRRSV infection [6, 7], and it was speculated that the IFN induced genes were predominantly expressed by the uninfected cells [7]. Here, another possibility is suggested that a certain amount of type-I IFN might be induced at the early stage of the infection before 5 dpi.
During HP-PRRSV infection, several aspects of the PAMs' function were under regulation, such as actin and tubulin cytoskeleton organization, exocytosis, protein degradation, protein folding, intracellular calcium and zinc homeostasis (Table 3). Increasing of intracellular zinc concentration impairs the replication of a variety of RNA viruses, including poliovirus, influenza virus, coronavirus, arterivirus, rhinovirus, and respiratory syncytial virus [40-42]. Recently, zinc ion has been reported to efficiently inhibit the RNA-synthesizing activity of the multiprotein replication and transcription complex of both SARS-coronavirus and equine arteritis virus [40]. Upregulation of SLC39A14 (also known as Zip14) (Table 3), a member of the SLC39 (Zip) family which transports zinc from the extracellular space or organellar lumen into the cytoplasm [43], might be a defense mechanism in PAMs during HP-PRRSV infection. Nevertheless, none of the SLC39 family genes was identified as a DE gene in a microarray assay of PAMs infected with PRRSV in vitro [8]. Furthermore, the expression of SLC39A7, another member of the SLC39 family, was downregulated in the lungs of Landrace×Yorkshire crossbred piglets at 7 dpi following HP-PRRSV infection [7].
It has been shown in this study, that modulated inflammatory reaction, with a few proinflammatory cytokines upregulated (CCL2, CCL4L and its receptor CCR5, and CSF1) (Table 4), might contribute to the mild regional lung lesion observed at 5 and 7 dpi (Figure 1C and D). Besides, the complement system is one of the key players in the defense against infections. However, excessive activation of the complement can also exaggerate the disease induced by viral or bacterial infection. In 2009, a new H1N1 influenza A virus caused severe disease in naive middle-aged human individuals with preexisting immunity against seasonal strains, and this disease is reported to be induced through high titers of low-avidity nonprotective antibody and immune complex-mediated complement activation in the respiratory tract [44]. Excessive complement activation can contribute to organ damage in combination with the cytokine storm in the later stages of sepsis caused by bacterial infection [45]. It is reported that blocking complement activation can ameliorate hepatic inflammation mediated by the hepatitis C virus core protein [46]. Likewise, inhibition of complement with a potent C3 inhibitor (compstatin) in a baboon model of late-stage sepsis markedly improves organ preservation and other clinical parameters [47]. As it has been shown here, inhibition of the complement system might also be a contributor to the mild regional lung damage during HP-PRRSV infection. Interestingly, infection of HP-PRRSV in six-week-old crossbred weaned pigs (Landrace × Yorkshire) induces complement activation accompanied by severe lung damage [7].
Conclusions
In summary, the data presented in this study suggested that during infection with HP-PRRSV Tongcheng piglets exhibited typical clinical signs, but displayed mild regional lung damage at 5 and 7 dpi. Microarray analysis revealed that HP-PRRSV infection has affected PAMs in vivo in expression of the important genes involved in cytoskeleton and exocytosis organization, protein degradation and folding, intracellular calcium and zinc homeostasis. Several potential antiviral strategies might be employed in PAMs, including upregulating IFN-induced genes and increasing intracellular zinc ion concentration. Furthermore, inhibition of the complement system likely attenuated the lung damage during HP-PRRSV infection. This system analysis could lead to a better understanding of the HP-PRRSV-host interaction, and to the identification of novel antiviral therapies and identifying genetic components for swine tolerance/susceptibility to HP-PRRS.
Supplementary Material
Supplementary Table 112,775 transcripts (53% of all probesets) expressed in infected and non-infected PAMs. TC: Tongcheng.
Differentially expressed (DE) genes. 321 DE genes identified under the threshold of fold change (FC) of 1.5 or greater and a false discovery rate (FDR) of approximately 0.5%.
Biological process Gene Ontology (GO) analysis of 166 differentially expressed genes. Many categories shared the same transcripts.
Acknowledgements
We would like to thank Tinghua Huang and Lijie Su in our lab for their help with microarray data analysis. We are grateful to Dr. Xiao Zhang (Uppsala University, Sweden) for valuable discussions and improving the manuscript. We thank our lab members for sample collections. The study is supported by Major Program of Natural Science Foundation of Hubei, Doctoral Fund of Ministry of Education of China (20100146110021), Fundamental Research Funds for the Central Universities (2011PY053), National Key Project for Breeding of New Transgenic Varieties (2009ZX08012-015B), and the Swedish Medical Research Council (K2009-67X-21128-01-3).
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Lewis CR, Torremorell M, Galina-Pantoja L, Bishop SC. Genetic parameters for performance traits in commercial sows estimated before and after an outbreak of porcine reproductive and respiratory syndrome. J Anim Sci. 2009;87:876-84
2. Thanawongnuwech R, Halbur PG, Andrews JJ. Immunohistochemical detection of porcine reproductive and respiratory syndrome virus antigen in neurovascular lesions. J Vet Diagn Invest. 1997;9:334-7
3. Murtaugh MP, Xiao Z, Zuckermann F. Immunological responses of swine to porcine reproductive and respiratory syndrome virus infection. Viral Immunol. 2002;15:533-47
4. Cheon DS, Chae C. Distribution of a Korean strain of porcine reproductive and respiratory syndrome virus in experimentally infected pigs, as demonstrated immunohistochemically and by in-situ hybridization. J Comp Pathol. 1999;120:79-88
5. Van Gucht S, Labarque G, Van Reeth K. The combination of PRRS virus and bacterial endotoxin as a model for multifactorial respiratory disease in pigs. Vet Immunol Immunopathol. 2004;102:165-78
6. Xiao S, Jia J, Mo D, Wang Q, Qin L, He Z, Zhao X, Huang Y, Li A, Yu J, Niu Y, Liu X, Chen Y. Understanding PRRSV infection in porcine lung based on genome-wide transcriptome response identified by deep sequencing. PLoS One. 2010;5:e11377
7. Xiao S, Mo D, Wang Q, Jia J, Qin L, Yu X, Niu Y, Zhao X, Liu X, Chen Y. Aberrant host immune response induced by highly virulent PRRSV identified by digital gene expression tag profiling. BMC Genomics. 2010;11:544
8. Genini S, Delputte PL, Malinverni R, Cecere M, Stella A, Nauwynck HJ, Giuffra E. Genome-wide transcriptional response of primary alveolar macrophages following infection with porcine reproductive and respiratory syndrome virus. J Gen Virol. 2008;89:2550-64
9. Miller LC, Harhay GP, Lager KM, Smith TP, Neill JD. Effect of porcine reproductive and respiratory syndrome virus on porcine alveolar macrophage function as determined using serial analysis of gene expression (SAGE). Dev Biol (Basel). 2008;132:169-74
10. Tian K, Yu X, Zhao T, Feng Y, Cao Z, Wang C, Hu Y, Chen X, Hu D, Tian X, Liu D, Zhang S, Deng X, Ding Y, Yang L, Zhang Y, Xiao H, Qiao M, Wang B, Hou L, Wang X, Yang X, Kang L, Sun M, Jin P, Wang S, Kitamura Y, Yan J, Gao GF. Emergence of fatal PRRSV variants: unparalleled outbreaks of atypical PRRS in China and molecular dissection of the unique hallmark. PLoS ONE. 2007;2:e526
11. Li B, Xiao S, Wang Y, Xu S, Jiang Y, Chen H, Fang L. Immunogenicity of the highly pathogenic porcine reproductive and respiratory syndrome virus GP5 protein encoded by a synthetic ORF5 gene. Vaccine. 2009;27:1957-63
12. Halbur PG, Paul PS, Meng XJ, Lum MA, Andrews JJ, Rathje JA. Comparative pathogenicity of nine US porcine reproductive and respiratory syndrome virus (PRRSV) isolates in a five-week-old cesarean-derived, colostrum-deprived pig model. J Vet Diagn Invest. 1996;8:11-20
13. Halbur PG, Paul PS, Frey ML, Landgraf J, Eernisse K, Meng XJ, Lum MA, Andrews JJ, Rathje JA. Comparison of the pathogenicity of two US porcine reproductive and respiratory syndrome virus isolates with that of the Lelystad virus. Vet Pathol. 1995;32:648-60
14. Zuckermann FA, Garcia EA, Luque ID, Christopher-Hennings J, Doster A, Brito M, Osorio F. Assessment of the efficacy of commercial porcine reproductive and respiratory syndrome virus (PRRSV) vaccines based on measurement of serologic response, frequency of gamma-IFN-producing cells and virological parameters of protection upon challenge. Vet Microbiol. 2007;123:69-85
15. Chen H, Li C, Fang M, Zhu M, Li X, Zhou R, Li K, Zhao S. Understanding Haemophilus parasuis infection in porcine spleen through a transcriptomics approach. BMC Genomics. 2009;10:64
16. Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185-93
17. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863-8
18. Dennis GJr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3
19. Tsai S, Cassady JP, Freking BA, Nonneman DJ, Rohrer GA, Piedrahita JA. Annotation of the Affymetrix porcine genome microarray. Anim Genet. 2006;37:423-4
20. Flori L, Rogel-Gaillard C, Cochet M, Lemonnier G, Hugot K, Chardon P, Robin S, Lefevre F. Transcriptomic analysis of the dialogue between Pseudorabies virus and porcine epithelial cells during infection. BMC Genomics. 2008;9:123
21. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-8
22. Misinzo GM, Delputte PL, Nauwynck HJ. Involvement of proteases in porcine reproductive and respiratory syndrome virus uncoating upon internalization in primary macrophages. Vet Res. 2008;39:55
23. Hinton A, Bond S, Forgac M. V-ATPase functions in normal and disease processes. Pflugers Arch. 2009;457:589-98
24. Carty M, Goodbody R, Schroder M, Stack J, Moynagh PN, Bowie AG. The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat Immunol. 2006;7:1074-81
25. Peng J, Yuan Q, Lin B, Panneerselvam P, Wang X, Luan XL, Lim SK, Leung BP, Ho B, Ding JL. SARM inhibits both TRIF- and MyD88-mediated AP-1 activation. Eur J Immunol. 2010;40:1738-47
26. El Kasmi KC, Smith AM, Williams L, Neale G, Panopoulos AD, Watowich SS, Hacker H, Foxwell BM, Murray PJ. Cutting edge: A transcriptional repressor and corepressor induced by the STAT3-regulated anti-inflammatory signaling pathway. J Immunol. 2007;179:7215-9
27. Strebovsky J, Walker P, Lang R, Dalpke AH. Suppressor of cytokine signaling 1 (SOCS1) limits NF{kappa}B signaling by decreasing p65 stability within the cell nucleus. FASEB J. 2011Mar;25(3):863-74
28. Wang X, Hinson ER, Cresswell P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe. 2007;2:96-105
29. Futatsumori M, Kasai K, Takatsu H, Shin HW, Nakayama K. Identification and characterization of novel isoforms of COP I subunits. J Biochem. 2000;128:793-801
30. Stankewich MC, Stabach PR, Morrow JS. Human Sec31B: a family of new mammalian orthologues of yeast Sec31p that associate with the COPII coat. J Cell Sci. 2006;119:958-69
31. Taylor KM, Morgan HE, Johnson A, Nicholson RI. Structure-function analysis of a novel member of the LIV-1 subfamily of zinc transporters, ZIP14. FEBS Lett. 2005;579:427-32
32. Quinn BJ, Welch EJ, Kim AC, Lokuta MA, Huttenlocher A, Khan AA, Kuchay SM, Chishti AH. Erythrocyte scaffolding protein p55/MPP1 functions as an essential regulator of neutrophil polarity. Proc Natl Acad Sci U S A. 2009;106:19842-7
33. Kowalska MA, Rauova L, Poncz M. Role of the platelet chemokine platelet factor 4 (PF4) in hemostasis and thrombosis. Thromb Res. 2010;125:292-6
34. Wang YS, Liao KW, Chen MF, Huang YC, Chu RM, Chi KH. Canine CXCL7 and its functional expression in dendritic cells undergoing maturation. Vet Immunol Immunopathol. 2010;135:128-36
35. Patel D, Nan Y, Shen M, Ritthipichai K, Zhu X, Zhang YJ. Porcine reproductive and respiratory syndrome virus inhibits type I interferon signaling by blocking STAT1/STAT2 nuclear translocation. J Virol. 2010;84:11045-55
36. Shi X, Wang L, Zhi Y, Xing G, Zhao D, Deng R, Zhang G. Porcine reproductive and respiratory syndrome virus (PRRSV) could be sensed by professional beta interferon-producing system and had mechanisms to inhibit this action in MARC-145 cells. Virus Res. 2010;153:151-6
37. Loving CL, Brockmeier SL, Sacco RE. Differential type I interferon activation and susceptibility of dendritic cell populations to porcine arterivirus. Immunology. 2007;120:217-29
38. Chung HK, Lee JH, Kim SH, Chae C. Expression of interferon-alpha and Mx1 protein in pigs acutely infected with porcine reproductive and respiratory syndrome virus (PRRSV). J Comp Pathol. 2004;130:299-305
39. Albina E, Carrat C, Charley B. Interferon-alpha response to swine arterivirus (PoAV), the porcine reproductive and respiratory syndrome virus. J Interferon Cytokine Res. 1998;18:485-90
40. Te Velthuis AJ, van den Worm SH, Sims AC, Baric RS, Snijder EJ, van Hemert MJ. Zn inhibits coronavirus and arterivirus RNA polymerase activity in vitro and zinc ionophores block the replication of these viruses in cell culture. PLoS Pathog. 2010;6:e1001176
41. Korant BD, Kauer JC, Butterworth BE. Zinc ions inhibit replication of rhinoviruses. Nature. 1974;248:588-90
42. Suara RO, Crowe JEJr. Effect of zinc salts on respiratory syncytial virus replication. Antimicrob Agents Chemother. 2004;48:783-90
43. Lazarczyk M, Favre M. Role of Zn2+ ions in host-virus interactions. J Virol. 2008;82:11486-94
44. Monsalvo AC, Batalle JP, Lopez MF, Krause JC, Klemenc J, Hernandez JZ, Maskin B, Bugna J, Rubinstein C, Aguilar L, Dalurzo L, Libster R, Savy V, Baumeister E, Cabral G, Font J, Solari L, Weller KP, Johnson J, Echavarria M, Edwards KM, Chappell JD, Crowe JEJr, Williams JV, Melendi GA, Polack FP. Severe pandemic 2009 H1N1 influenza disease due to pathogenic immune complexes. Nat Med. 2010 [Epub ahead of print]
45. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785-97
46. Chang ML, Yeh CT, Lin DY, Ho YP, Hsu CM, Bissell DM. Hepatic inflammation mediated by hepatitis C virus core protein is ameliorated by blocking complement activation. BMC Med Genomics. 2009;2:51
47. Silasi-Mansat R, Zhu H, Popescu NI, Peer G, Sfyroera G, Magotti P, Ivanciu L, Lupu C, Mollnes TE, Taylor FB, Kinasewitz G, Lambris JD, Lupu F. Complement inhibition decreases the procoagulant response and confers organ protection in a baboon model of Escherichia coli sepsis. Blood. 2010;116:1002-10
48. Van Gorp H, Van Breedam W, Delputte PL, Nauwynck HJ. The porcine reproductive and respiratory syndrome virus requires trafficking through CD163-positive early endosomes, but not late endosomes, for productive infection. Arch Virol. 2009;154:1939-43
Author contact
![]() Corresponding author: liubanghzau.edu.cn; Tel: +86 27 87284140; Fax: +86 27 87280408
Corresponding author: liubanghzau.edu.cn; Tel: +86 27 87284140; Fax: +86 27 87280408