Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(4):242-250. doi:10.7150/ijbs.3.242 This issue Cite
Research Paper
Silencing of the Pink1 Gene Expression by Conditional RNAi Does Not Induce Dopaminergic Neuron Death in Mice
Hongxia Zhou1, Björn H Falkenburger2, Jörg B Schulz2, Kim Tieu3, Zuoshang Xu4, Xu Gang Xia1 ![]()
1. Department of Pathology, Anatomy & Cell Biology, Thomas Jefferson University, 1020 Locust Avenue, Philadelphia, PA 19107, USA
2. Department of Neurodegeneration and Restorative Research, DFG Research Center Molecular Physiology of the Brain and Center of Neurological Disease, University of Göttingen, Waldweg 33, 37073 Göttingen, Germany
3. Department of Environmental Medicine and Center for Aging and Developmental Biology, University of Rochester, School of Medicine and Dentistry, 575 Elmwood Avenue, Rochester, NY 14642, USA
4. Department of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School, 364 Plantation St, Worcester, MA 01605, USA
Received 2006-12-31; Accepted 2007-3-2; Published 2007-3-5
Abstract
Transgenic RNAi, an alternative to the gene knockout approach, can induce hypomorphic phenotypes that resemble those of the gene knockout in mice. Conditional transgenic RNAi is an attractive choice of method for reverse genetics in vivo because it can achieve temporal and spatial silencing of targeted genes. Pol III promoters such as U6 are widely used to drive the expression of RNAi transgenes in animals. Tested in transgenic mice, a Cre-loxP inducible U6 promoter drove the broad expression of an shRNA against the Pink1 gene whose loss-of-functional mutations cause one form of familial Parkinson's disease. The expression of the shRNA was tightly regulated and, when induced, silenced the Pink1 gene product by more than 95% in mouse brain. However, these mice did not develop dopaminergic neurodegeneration, suggesting that silencing of the Pink1 gene expression from embryo in mice is insufficient to cause similar biochemical or morphological changes that are observed in Parkinson's disease. The results demonstrate that silencing of the PINK1 gene does not induce a reliable mouse model for Parkinson's disease, but that technically the inducible U6 promoter is useful for conditional RNAi in vivo.
Keywords: RNAi, transgenic RNAi, U6 promoter, Parkinson disease, PINK1, mice
1. Introduction
RNA interference (RNAi) is widely used to silence gene expression in the study of gene functions and in the development of therapeutic strategies [1-3]. RNAi is induced by short double-stranded RNA called small interfering RNA (siRNA), of which one strand (guide strand) is incorporated into RNA-Induced Silencing Complex (RISC) and directs localization and degradation of targeted RNA molecules with complementary sequence to the siRNA guide strand [4-6]. Sustained RNAi can be achieved by introducing a gene-based construct that synthesizes short hairpin RNA (shRNA) from either RNA polymerase II or III (Pol II or III) promoters [2, 3]. Pol III promoters such as U6 or H1 offer advantages over Pol II promoters, because the compact sequence of Pol III promoters is easier to handle and their ubiquitous expression pattern allows for broad silencing of a target gene [7-9]. An shRNA transcript synthesized from a Pol III promoter is exported by Exportin-5 to the cytoplasm where it is processed by Dicer to form siRNA [10, 11], the final and functional product of shRNA. If an shRNA-expressing construct is integrated into the genome, it becomes a sustainable source of siRNA and produces sustained RNAi effects, an approach that is called transgenic RNAi. This approach has been demonstrated to successfully reproduce phenotypes of the gene knockouts [12-14].
Transgenic RNAi is a convenient alternative to the gene knockout approach for the following reasons: it can avoid the use of embryonic stem cells; it enables the generation of varying degrees of hypomorphic phenotypes because RNAi-mediated silencing is dose-dependent of siRNA; and it can be applied to mammalian species other than mice (such as rats) in which gene knockout has not yet been established [3, 12]. When a constitutive promoter is used to drive shRNA expression, the dominant nature of RNAi limits its use to target some critical genes whose loss results in premature death or infertility. To overcome this drawback, a variety of inducible promoters such as Cre-loxP-regulated U6 have been created and tested in transgenic studies [13, 15, 16]. Several strategies are available to create Cre-LoxP-regulated U6 promoters: the first is to place the loxP site between the distal sequence element (DSE) and the proximal sequence element (PSE) of the promoter [13, 16]; the second is to use the loxP site as a loop within the shRNA structure [17]; and the third is to modify the TATA box of the promoter to introduce a mutant loxP site [15, 18]. Our previous studies tested the first strategy and found a leakage of the promoter in transgenic mice [16]. The second strategy compromises the efficiency of shRNA processing because the shRNA is added with an excessive length of the loop sequence [19]. The third strategy has been developed independently by different groups [15, 18], and was reported to tightly regulate shRNA expression in cell culture and in mouse embryos. Here we tested the third strategy in adult mice in which the PTEN-induced putative kinase 1 (PINK1) gene, a Parkinson disease (PD)-associated gene, was targeted.
Most PD cases are sporadic, but some rare forms are caused by mutations in individual genes. PINK1 is a mitochondrial protein which is thought to cause PD by loss-of-functional mutations and whose depletion leads to mitochondrial dysfunction in drosophila [20, 21]. PINK1 knockout mice have not been reported. Using transgenic RNAi approach, we generated conditional PINK1-silenced mice. In these transgenic mice, the expression of the PINK1 shRNA was tightly regulated by the Cre-loxP system and activation of the RNAi transgene resulted in silenced expression of the Pink1 gene in various tissues and to less than 5% of the normal levels in the central nerve system. Silencing of the PINK1 gene expression did not cause a loss of the dopaminergic neurons in the PINK1 RNAi transgenic mice.
2. Materials and Methods
Plasmid construction
The coding sequence of the mouse PINK1 gene was copied by PCR from a cDNA pool that was reverse-transcribed from the cellular RNA of mouse brain. The PINK1 open reading frame (ORF) was cloned downstream of the ORF of enhanced green fluorescence protein (EGFP; Figure 1A) in the vector pEGFP-N1 (Clonetech).
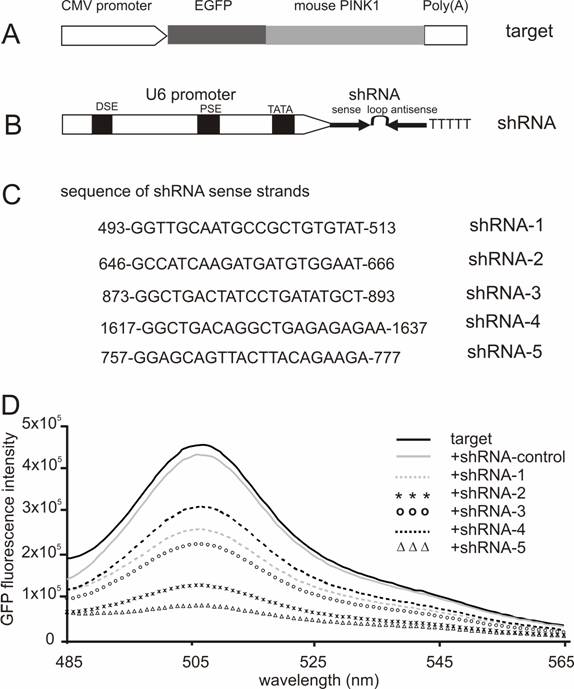
Screening for effective shRNA against the mouse Pink1 gene. (A) A mouse Pink1 gene was inserted at the C-terminal and in the frame of the EGFP gene that was driven by the CMV promoter; the fusion protein served as an indicator for the RNAi-mediated knockdown of Pink1 gene expression. (B) The native mouse U6 promoter was used to drive the expression of PINK1-shRNA gene that consisted of an shRNA sense strand, the loop sequence TTCAAGAGA, and an shRNA antisense strand. (C) The shRNA targeting sequences on mouse PINK1 mRNA were shown when the translation start-site was set at 1. (D) The fluorescent intensity of GFP protein was measured in the lysate of cells that were transfected with a combination of EGFP-PINK1 expression vector (A) with either the PINK1-shRNA or an shRNA-control vector that does not target any gene.
U6-shRNA vectors were constructed as described earlier [16, 22]. Briefly, single-stranded DNA oligonucleotides were chemically synthesized and annealed to form double-stranded DNA that consisted of shRNA sense sequence, the loop sequence TTCAAGAGA, shRNA antisense sequence, and a string of six thymidine residues which served as transcription-terminating signal for RNA polymerase III promoters. The double-stranded DNA oligonucleotide flanked with restriction sites was cloned downstream of mouse U6 promoter in an expression vector. For conditional expression of shRNA, a Cre-loxP-inducible U6 promoter was generated by a similar strategy which mutated the sequence around TATA box of the U6 promoter to create a mutant loxP site [15, 18]. The U6 promoter was reversibly inactivated after a stuffer sequence of 1kb was inserted between the two mutant loxP sites (Figure 2A), but to be recovered by Cre-mediated excision of the stuffer sequence [23] (Figure 2B). The stuffer sequence contained multiple strings of thymidine along with the neomycin gene ORF and the SV40 poly (A) signal. All the plasmids were sequence-verified.
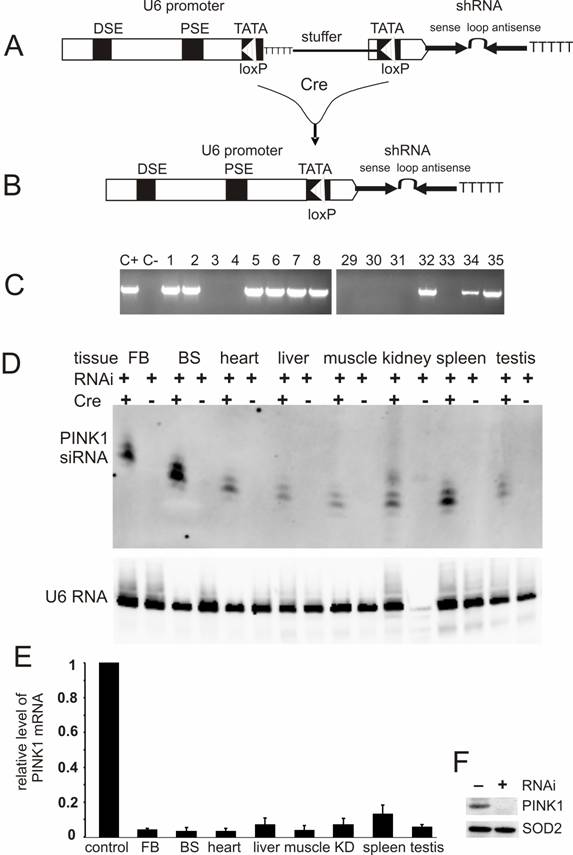
Conditional expression of PINK1-shRNA silenced PINK1 gene expression in the transgenic mice. (A) PINK1-RNAi transgenic mice were generated by pronuclear injection of a PINK1-shRNA construct under control of a Cre-loxP-inducible mouse U6 promoter that was temporally inactivated by insertion of a LoxP-flanked stuffer sequence at its TATA box. (B) After Cre-mediated excision of the stuffer sequence from the promoter region, the integrity of the U6 promoter was recovered in the double transgenic mice that carried both the Cre and PINK1-RNAi transgenes. (C) The RNAi transgene-positive founders were identified by PCR amplification of a fragment of the transgene DNA extracted from mouse tail. Positive control (C+): PCR amplification of the transgene construct; negative control (C-): PCR amplification of genomic DNA extracted from wildtype mouse tail. (D) The expression of the PINK1-RNAi transgene in mouse tissues (FB, forebrain; BS, brainstem) was measured with Northern blotting in the presence of an RNA probe complementary to the antisense strand of the PINK1-RNAi transgene. Each lane was loaded with 20µg of total cellular RNA that was extracted from the tissues of the Cre and PINK1-RNAi double transgenic (RNAi+/Cre+) or the PINK1-RNAi single transgenic mice (RNAi+/Cre-). Equal loading was controlled by probing the upper part of the same membrane with a mouse U6 RNA-specific probe. (E) The expression of PINK1 mRNA was examined with quantitative PCR, and the level of PINK1 mRNA was calculated relative to that of the housekeeping gene L17. The degrees of RNAi-mediated knockdown were expressed as a ratio of the relative level of PINK1 mRNA in the RNAi-transgenic tissue to that in non-transgenic tissue in which the relative level of PINK1 mRNA was set to 1 (control). Abbreviations are FB for forebrain, BS for brainstem, and KD for kidney. (F) Western blot measured the level of PINK1 protein in mitochondrial extract from mouse brain. Equal loading was examined by probing the same membrane with an antibody against the mitochondrial protein SOD2.
Cell culture and transfection
HEK293 cells were grown in DMEM medium supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100µg/ml streptomycin. The day before transfection, the cells (70-90% of confluence) were detached by trypsin treatment, plated onto 6-well plates, and cultured in 10% FBS-containing medium without antibiotics. In the absence of serum, the cells were transfected with the transfection reagent lipofectamine-2000 (Invitrogen) per manufacturer's instruction. FBS (10%) and antibiotics were added four hours after transfection. The growth medium was changed every 24 hours.
Fluorescence measurement
Cells were harvested by centrifugation and snap-frozen in liquid nitrogen at 28 hours after transfection. The cell pellet was lyzed in ice-cold reporter buffer (Promega) that was supplemented with protease inhibitors (complete, EDTA-free, 1 tablet/10 ml buffer; Roche). The lysate was cleared by centrifugation (14000 rpm) at 4°C for 10 minutes. Total protein in the supernatant was measured by BCA assay (Pierce; Rockville IL). Protein concentration of each sample was adjusted to 0.5 mg/ml with the reporter buffer. Fluorescence of green fluorescence protein (GFP) in 140 µl of samples was measured by fluorescence spectroscopy (Photon Technology International) with excitation at 460 nm and recording from 485 to 565 nm. The spectrum peak detected at 505 nm represented the fluorescence intensity of GFP. Fluorescence in untransfected lysate was measured as background and subtracted from measurements of the transfected lysate [22, 24].
Generation of RNAi transgenic mice
A PINK1-shRNA construct of 2.2kb was linearized by restriction digestion and purified from agarose gel (Figure 2A). The RNAi transgenic mice were generated by pronuclear injection of the transgene construct into fertilized eggs that were produced from the crossing of C57BL/6 with SJL mice. Transgene-positive founder and offspring were identified by PCR-magnification of a part of the transgene, a part which was across the promoter region and the stuffer sequence (forward primer: 5'-AGACTTGTGGGAGAAGCTCG-3'; reverse primer: 5'-GCCTCTTCATCGGGAATGC-3'). The promoter activity was recovered after Cre-mediated excision of the stuffer sequence, and the RNAi transgene was thus expressed. The RNAi-active mice that carried both the Cre and PINK1-shRNA transgenes were identified by PCR-magnification of a part of the RNAi transgene, a part that consisted of the promoter sequence and the sense sequence of the shRNA (forward primer: 5'-AGCCGATTGTCTGTTGTGC-3'; reverse primer: 5'- CTTCTGTAAGTAACTGCTCC-3'). The PINK1-RNAi active mice were backcrossed to FVB genomic background for at least five generations before they were used for experiments. Animal usage followed the NIH guideline and the protocol was approved by the Institutional Animal Care and Use Committees (IACUC) of the authors' institutions.
Northern blot
Mice were decapitated under deep anesthesia and their tissues were quickly dissected, snap-frozen in liquid nitrogen, and stored at -80ºC. Total cellular RNA was extracted from the frozen tissues with the reagent Trizol (Sigma). RNA samples (20 µg each) were separated on 12% polyacrylamide gels and transferred onto Hybond TM-N+ membranes (Amersham). After UV cross-linking, the membrane was cut into two parts: the low part was probed with an RNA probe complementary to the antisense strand of the mouse PINK1 shRNA; the upper part was probed with an RNA probe complementary to a part of mouse U6 RNA (the sequence of U6 RNA probe: 5'-TTCACGAATTTGCGTGTCATCCTTGCG-3'). RNA probes were synthesized by in vitro transcription with T7 RNA polymerase and labeled by addition of Digoxigenin-11-uridine-5'-triphosphate instead of uridine-5'-triphosphate during RNA synthesis (RNA probe synthesis kit: Roche). After probing with specific RNA probes, the membranes were incubated with anti-Digoxigenin-AP antibodies (Roche), and the signals were detected with CDP-star kit and documented on a Kodak Image Station.
Quantitative PCR
For determination of mRNA levels, total RNA isolated with Trizol reagent was further purified with RNA easy kit and digested on column with RNase-free DNase (Qiagen). One microgram of purified total RNA from each sample was reversely transcribed to cDNA with oligo-dT primer (RT kit, Invitrogen), and the resulting cDNA was measured by quantitative PCR with SYBR green kit per manufacturer's instruction (Qiagen). For quantitative PCR, the following primers were used at a concentration of 500nM: mouse PINK1 primers GCTTGCCAATCCCTTCTATG (forward) and CTCTCGCTGGAGCAGTGAC (reverse); OAS1 primers CGTGCTGCCAGCCTATGATTT (forward) and TTGGTTGGGCGACAGTTCAG (reverse); STAT1 primers GCTGGGCGTCTATCCTGTGGT (forward) and GCTCAGCTGGTCTGCGTTCA (reverse); and mouse L17 primers CGGTATAATGGTGGAGTTG (forward) and ACCCTTAAGTTCAGCGTTACT (reverse). Cycling conditions were 15 min at 95°C followed by 40 cycles of 15 seconds at 94°C, 30 seconds at 60°C, and 20 seconds at 72°C. Aliquots of the amplified products were separated on 3% agarose gels to ensure amplification of the specific products at the predicted length. The threshold cycle number (Ct) for an examined gene was normalized to the Ct for the housekeeping gene, namely ribosomal RNA L17. The relative mRNA level of the gene of interest was determined and expressed as a ratio of the mRNA level in the transgenic mouse tissue to that in the wildtype mouse tissues as described previously [16].
Since the PINK1 RNAi transgene is driven by mouse U6 promoter, the copy number of the RNAi transgene can be estimated by determination of the total copies of mouse U6 promoters in the endogenous U6 gene and in the PINK1 RNAi transgenes. The number of copies of the PINK1 RNAi transgenes was estimated by real-time PCR analysis of mouse tail DNA. The primers for the real-time PCR were designed for magnification of mouse U6 promoter: TCCGACGCCGCCATCTCTA (forward) and TATCGCACATTAAGCCTCTA (reverse). The Ct of the U6 promoter was normalized to the internal control glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which was amplified using primers CCACTCTTCCACCTTCGATG (forward) and TCCACCACCCTGTTGCTGTA (reverse). The copy number of the PINK1 RNAi transgene was calculated relative to the endogenous mouse U6 gene which is known two copies per genome [16].
Western blot
Mice were decapitated under deep anesthesia and their brains were dissected. Mouse brains were minced in a buffer (pH 7.4) that consisted of 10 mM Tris-HCl, 0.5 mM EDTA, 0.25 M sucrose, and a mixture of protease inhibitors (Sigma). The minced tissue was further homogenized with Dounce homogenizers for 20 strokes each with pestle A (clearance 0.12 mm) and pestle B (clearance 0.06 mm). Tissue debris, unbroken cells, and cell nuclei were removed by centrifugation at 1000 x g for 20 minutes; the supernatant was centrifuged at 16,000 g for 20 minutes to pellet gross mitochondria as described previously [25]. The mitochondrial pellets were homogenized in ice-cold lysis buffer that consisted of 0.4% NP-40, 0.2 mM Na3VO4, 20 mM HEPS (pH 7.9), and a cocktail of protease inhibitors (Sigma). Mitochondrial lysates were cleared by centrifugation, and protein content in the cleared lysate was determined by BCA assay. Equal amounts of protein were resolved on precast 4-20% gradient SDS-PAGE and blotted onto GeneScreen Plus membrane (Perkin Elmer). PINK1 protein was detected by incubation of the membrane with a rabbit anti-PINK1 antibody (Cayman Chemical: #10006283) and by signal development with Super Signal kit (Pierce). After detection of PINK1 protein, the membrane was stripped in an SDS buffer (2% SDS, 100 mM ß-mercaptoethanol, 62.5 mM Tris-HCl, pH 6.7) at 55 °C for 30 minutes and used for detecting immunoreactivity of the mitochondrial protein SOD2, which served as a loading control [14].
Immunostaining and cell counting
Mice were decapitated and their brains were removed and cut into two parts: the front section was used for dissection of the striatum over ice and the back section was submersion-fixed in 4% paraformaldehyde in 0.1 M phosphate-buffered saline at 4ºC. After fixation in 4% paraformaldehyde for 2 days, mouse brains were dehydrated in 30% sucrose and then cut into serial coronal sections (40 μm thick) on a Cryostat. Every third section through the entire midbrain was incubated with a rabbit antibody against tyrosine hydroxylase (TH; 1:1000; Pel-Freez), an enzyme that catalyzes the rate-limiting step in dopamine synthesis and is a reliable marker for viable dopaminergic neurons. After thorough washes, tissue sections were sequentially incubated with biotinylated goat anti-rabbit IgG (1:500; Vector Laboratories), and thereafter with peroxidase-conjugated avidin-biotin complex (ABC kit; Vector Laboratories). Bound antibodies were visualized by addition of diaminobenzidine (Vector Laboratories), and tissue sections were mounted in sequential order (rostral to caudal), dried and covered. The total number of TH-positive neurons in the substantia nigra pars compact (SNc) was estimated by stereological counting with a fractionator-based unbiased assay, as described earlier [26]. Therefore, the number of TH-positive neurons was independent of their size, shape or orientation; tissue shrinkage; or anatomical level.
HPLC measurement
Dissected mouse striata were homogenized in 0.1M perchloric acid by sonication at 4ºC. Tissue lysate was cleared by centrifugation at 4ºC, and the supernatant was collected for measurement of dopamine and its metabolites by electrochemical detection using high performance liquid chromatography as described [27].
Behavior test
Mouse motor function was evaluated with an accelerating rotarod as described [16]. Mice were trained to stay on a rotating rotarod two days before performing the motor function test which was done monthly. Each animal was tested for 3 trials each time and the longest time that the mouse stayed on the rotating rod was used for analysis of motor function.
Statistical analysis
Data were expressed as means ± SEM values. The statistical significance was tested with ANOVA followed by Tukey's post hoc test to compare group means. In all analyses, the null hypothesis was rejected at a level of 0.05.
3. Results
Selection of effective PINK1-shRNA
RNAi-mediated inhibition is dose-dependent of the RNAi trigger siRNA, but the maximal inhibition depends on the potency of the RNAi trigger [14, 28]. Besides its gene-specific inhibition, RNAi may also cause side-effects or toxicities which are dependent on sequence and dose of siRNA or shRNA used [29-31]. To achieve maximal RNAi in vivo without unexpected effects, a potent shRNA should be selected and used for pronuclear injection. Five shRNA-targeting sequences were chosen from the coding sequence of mouse Pink1 gene, and the RNAi efficacy of each shRNA was tested in HEK293 cells. After cotransfection with the GFP-tagged Pink1 mini-gene, the intensity of GFP fluorescence was determined and used as an indicator for target gene expression. Although a control shRNA (sense sequence: CCAGACTTAGCTGCTGTATCGGCT) which did not target any gene failed to induce any inhibition, all five PINK1-shRNA silenced their target gene expression by different degrees from 50% to 85%, indicating that the RNAi effect was sequence-dependent of the PINK1-shRNA (Figure 1). These two shRNAs, #2 and #5, gave the best inhibition when the shRNA was used at a low ratio to its target by 1:2. In transient transfection, an shRNA that is able to inhibit its target expression by 80% can be defined effective [14, 22]. Thereby, we chose PINK1-shRNA-5 for the subsequent transgenic study.
Expression of PINK1 shRNA from a Cre-loxP-inducible U6 promoter was tightly regulated
For conditional RNAi, the effective PINK1-shRNA was expressed from a Cre-loxP inducible U6 promoter which was reversibly inactivated by insertion of a loxP-flanked stuffer sequence at the TATA box [15, 18]. By pronuclear injection, 54 transgenic founders were generated and nine were identified transgene-positive (Figure 2C). Five positive lines gave offspring that carried the RNAi transgene in germline (data not shown). None of the RNAi transgenic mice expressed the transgene before they were mated with a Cre-transgenic mouse, indicating no detectable leakage of the Cre-loxP-regulatable U6 promoter. U6 promoter activity was expected to be restored in the double transgenic mice that were generated by the crossing of PINK1-RNAi transgenic mice with CMV-Cre transgenic mice because the Cre enzyme would mediate excision of the stuffer sequence and recover the U6 promoter activity. Non-excision or incomplete excision was identified by PCR-detection of the stuffer sequence; whereas, complete excision was identified by a lack of the stuffer sequence and a recovery of the distance between the U6 promoter and the shRNA upon PCR detection. Cre-mediated excision was complete in every line except line #35, which displayed incomplete excision of the stuffer sequence by Cre activity (data not shown). Nevertheless, the product of RNAi transgene was detected in different tissues of the double transgenic mice from line #35 as well as line #8. The CMV-Cre transgenic mice express Cre recombinase ubiquitously [32], and thus, the PINK1 RNAi transgene can be activated in every tissue. RNAi transgene product was detected by Northern blot in all tissues (Figure 2D, E). Line #8 expressed the RNAi transgene at a higher level than line #35 and was used for further experiments. No leakage in shRNA expression was detected before Cre induction (Figure 2D). By quantitative PCR, we measured the copy number of the PINK1 RNAi transgenes for these two expressing lines: #8 and #35. Initially, the line-8 carries 3 copies of the quiescent PINK1 RNAi transgene and the line-35 carries 4 copies of the transgene. After crossing with the CMV-Cre transgenic mice, the PINK1 RNAi transgene was activated by Cre-mediated excision of the intervening sequence from the promoter region. In the Cre and PINK1-RNAi double transgenic mice, the copy number was reduced to one copy for the line-8, but to 2 copies for the line-35.
Conditional RNAi silenced Pink1 gene expression but did not cause dopaminergic death
Broad expression of PINK1 shRNA resulted in a significant knockdown of the PINK1 mRNA in the central nervous system, heart, liver, kidney, muscle, spleen, and testis (Figure 2E). The level of the PINK1 mRNA was reduced to the detection limit by PCR in mouse brainstem where dopaminergic neurons reside, and the level of PINK1 protein was below detection threshold of Western blot in the mitochondrial proteins from mouse brain (Figure 2F).
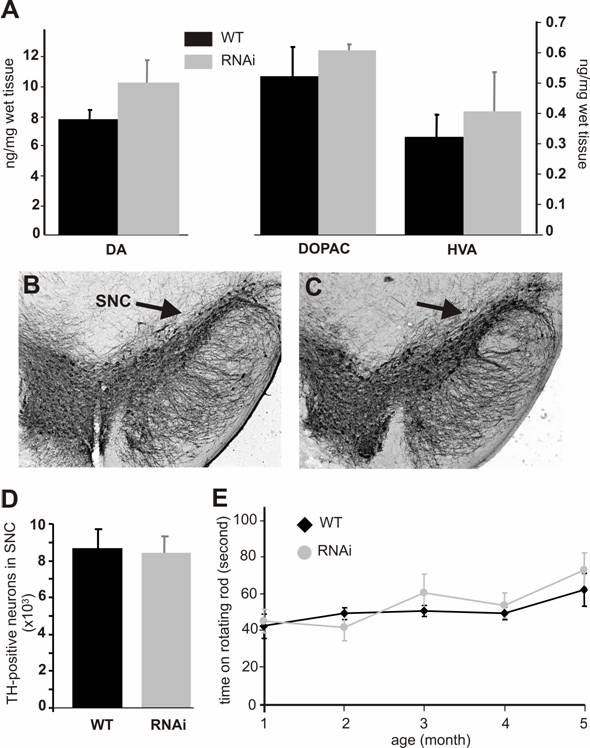
Both PINK1 RNAi-inactive (before Cre-mediated excision) and RNAi-active (after Cre-mediated excision) mice did not present gross abnormalities. PINK1 is associated with familial Parkinson's disease that is characterized by progressive degeneration of the dopaminergic neurons in SNc and the dopaminergic terminals in the striatum. To determine whether these characteristics were recapitulated in PINK1-silenced mice, we examined the TH-positive neurons in the SNc. No difference in cell and tissue structure was detected (Figure 3B, C). Stereological counting of the TH-positive neurons in the SNc did not reveal a significant difference between the wild type and the PINK1-silenced mice at age of 6 months (Figure 3D) or 1 year (data not shown). Measurements of dopamine and its metabolites showed a trend – though not statistically significant – for increased concentrations in the striatum (Fig. 3A). Consistent with the normal level of striatal dopamine, the motor function of PINK1-silenced mice was also normal when tested on a rotorod (Figure 3E).
Silencing of the PINK1 gene expression does not cause dopaminergic neuron death. (A) The striatal contents of the neurotransmitter dopamine (DA) and its metabolites DOPAC and HVA were measured with HPLC in the tissues of the RNAi-activated mice (RNAi) or its wildtype littermate (WT) at age of 6 months. Representative photos of midbrain section immunostained for TH were taken from the wildtype (B) or the PINK1-RNAi active mice (C). Arrows point to substantia nigra pars compacta (SNc). (D) The total number of TH-positive neurons in the SNc was counted stereologically. (E) Mouse motor activity was measured on rotating rod. Abbreviations are WT for wildtype and RNAi for RNAi-transgenic mice. Data are mean ± SEM (n = 4-6).
Long-term expression of PINK1 shRNA did not cause dsRNA-dependent interferon response
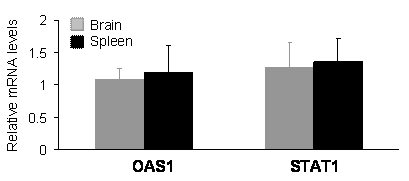
In mammals, expression of shRNA may cause dsRNA-dependent interferon response which is dependent on the sequence length and content of shRNA [29-31]. An shRNA longer than 30 nucleotides may activate interferon pathways; whereas, a shorter one may not. The commonly used shRNA of 21-27 nucleotides can avoid interferon response in most situations; however, some short shRNA can still stimulate the interferon signaling pathway [29-31]. The results of gene silencing may be complicated by potential interferon response. To exclude this possibility, we examined the expression of oligoadenylate synthetase 1 (OSA1) and signal transducer and activator of transcription 1 (STAT1), which are known to be induced by some shRNA [29-31]. Real-time PCR analysis failed to detect an upregulation of the OSA1 and STAT1 genes in the brain and spleen tissues of PINK1 RNAi transgenic mice (Figure 4), indicating that expression of the PINK1 shRNA transgene did not induce significant interferon response.
Expression of the PINK1-RNAi transgene did not upregulate the expression of OSA1 and STAT1 genes in the transgenic mice. Levels of the mRNA for each gene were determined by quantitative PCR. The levels of OAS1 and STAT1 mRNA were normalized to that of GAPDH mRNA, and the relative mRNA level in the RNAi-transgenic tissues was expressed as a ratio to that in wildtype tissues. Data are mean ± SEM (n = 4).
4. Discussion
We tested a Cre-loxP-regulatable U6 promoter in RNAi transgenic mice. The results showed that this promoter is fully regulatable in that it was completely inactive before induction, but upon Cre induction, it drove the expression of the shRNA ubiquitously. Silencing of the Pink1 gene expression did not induce Parkinson's disease phenotypes, including a loss of the dopaminergic neurons in the SNc and the neurotransmitter dopamine in the striatum.
Transgenic RNAi is an attractive approach for reverse genetics in mice because it has certain advantages. It is faster and cheaper as compared with gene knockout approach by homologous recombination. It can be applied virtually in all mammalian species as exemplified by its capability of silencing genes in mice, rats and goat [12-14, 33-36]. Inducible RNAi based on the Cre-loxP system has also been developed in transgenic mice, and it enables the investigator to control gene silencing spatially and temporally [13, 16, 37]. One design of Cre-loxP inducible U6 promoter is to increase the distance between DSE and PSE elements of the promoter [16]. A shortcoming of this design is a certain level of leakage expression of shRNA before Cre induction. This is not surprising because even a DSE-deleted U6 promoter still has some promoter activity [38, 39]. By using a modified loxP sequence that contains a TATA box and by insertion of a stuffer sequence between PSE and TATA box, the expression of shRNA by the modified U6 promoter is interrupted [38, 39]. Previous studies have shown an absence of shRNA synthesis in oocytes and T cells for the regulated promoter [38, 39]. We applied this U6 promoter to drive the PINK1 RNAi transgene and demonstrate the absence of any leakage of the transgene expression in all tissues of the transgenic mice and a functional recovery of the promoter upon Cre induction. Thus, this Cre-loxP regulated U6 promoter is reliable for reverse genetics in vivo.
Transgenic RNAi has been demonstrated capable of silencing gene expression in transgenic mice and inducing phenotypes of the gene knockout. In transgenic mice, an shRNA against Id4 gene induces developmental defects in kidney [12]; silencing of Fgfr2 gene causes embryonic lethality [13]; inhibition of Msy2 gene expression in oocytes results in reduced fertility [33]; and silencing of Sod2 gene by transgenic RNAi leads to SOD2 dysfunction that results in severe consequences resembling the phenotypes of Sod2 gene knockout [14]. Transgenic RNAi can recapitulate gene knockout phenotypes by RNAi-mediated continuous degradation of targeted gene product, namely, mRNA. In the PINK1 RNAi transgenic mice, the level of PINK1 mRNA was less than 5% of the mRNA in wildtype littermates. Drastic degradation of PINK1 mRNA led to invisibility of PINK1 protein on Western blot. However, silencing of PINK1 gene did not induce dopaminergic death. This may not be surprising because knockout of two other Parkinson's disease-linked genes, Parkin and DJ-1, fails to reproduce Parkinson's disease phenotypes in mice [40-42]. Deletion of either the Parkin or DJ-1 genes by gene knockout approach does not cause dopaminergic neuron death [43-49]. Silencing of the PINK1 gene is insufficient to induce dopaminergic death in mice, though deletion of the PINK1 gene causes severe phenotypes in Drosophila [20, 21]. This could be interpreted as a difference in animal species. Similarly, deletion of the Parkin gene in Drosophila causes severe phenotypes resembling the PINK1 knockout fly [50], but no loss of dopaminergic neurons is observed in Parkin knockout mice [44, 46]. Our results suggest that a lack of PINK1 gene function is insufficient to cause dopaminergic neuron death in mice and indicate that this version of Cre-loxP regulated U6 promoter is reliable for conditional gene silencing in vivo.
Acknowledgements
We thank Dr. Steve Jones and the University of Massachusetts Medical School transgenic core for pronuclear injection. This work is supported by grants from NIH/NINDS (RO1NS048145) and NIH/NIA (R21AG023808) to Zuoshang Xu. BF and JBS are supported by funds from the German Research Foundation to the CMPB. The contents of this report are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Competing interests
The authors declare no conflicts of interest.
References
1. Heck S, Qian X, Velleca M. Genetically engineered mouse models for drug discovery: new chemical genetic approaches. Curr Drug Discov Technol. 2004;1(1):13-26
2. Spankuch B, Strebhardt K. RNA interference-based gene silencing in mice: the development of a novel therapeutical strategy. Curr Pharm Des. 2005;11(26):3405-3419
3. Xia XG, Zhou H, Xu Z. Transgenic RNAi: Accelerating and expanding reverse genetics in mammals. Transgenic Res. 2006;15(3):271-275
4. Wiznerowicz M, Szulc J, Trono D. Tuning silence: conditional systems for RNA interference. Nat Methods. 2006;3(9):682-688
5. Tijsterman M, Plasterk RH. Dicers at RISC; the mechanism of RNAi. Cell. 2004;117(1):1-3
6. Hannon GJ, Rossi JJ. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431(7006):371-378
7. Paule MR, White RJ. Survey and summary: transcription by RNA polymerases I and III. Nucleic Acids Res. 2000;28(6):1283-1298
8. Jensen RC, Wang Y, Hardin SB, Stumph WE. The proximal sequence element (PSE) plays a major role in establishing the RNA polymerase specificity of Drosophila U-snRNA genes. Nucleic Acids Res. 1998;26(2):616-622
9. Seibler J. et al. Single copy shRNA configuration for ubiquitous gene knockdown in mice. Nucleic Acids Res. 2005;33(7):e67
10. Ohrt T. et al. In situ fluorescence analysis demonstrates active siRNA exclusion from the nucleus by Exportin 5. Nucleic Acids Res. 2006;34(5):1369-1380
11. Yi R. et al. Overexpression of exportin 5 enhances RNA interference mediated by short hairpin RNAs and microRNAs. RNA. 2005;11(2):220-226
12. Peng S, York JP, Zhang P. A transgenic approach for RNA interference-based genetic screening in mice. Proc Natl Acad Sci U S A. 2006;103(7):2252-2256
13. Coumoul X. et al. Conditional knockdown of Fgfr2 in mice using Cre-LoxP induced RNA interference. Nucleic Acids Res. 2005;33(11):E102
14. Xia XG. et al. Pol II-expressed shRNA knocks down Sod2 gene expression and causes phenotypes of the gene knockout in mice. PLoS Genet. 2006;2(1):27
15. Ventura A. et al. Cre-lox-regulated conditional RNA interference from transgenes. Proc Natl Acad Sci U S A. 2004;101(28):10380-10385
16. Xia XG, Zhou H, Huang Y, Xu Z. Allele-specific RNAi selectively silences mutant SOD1 and achieves significant therapeutic benefit in vivo. Neurobiol Dis. 2006;23(3):578-586
17. Kasim V, Miyagishi M, Taira K. Control of siRNA expression using the Cre-loxP recombination system. Nucleic Acids Res. 2004;32(7):e66
18. Tiscornia G, Tergaonkar V, Galimi F, Verma IM. CRE recombinase-inducible RNA interference mediated by lentiviral vectors. Proc Natl Acad Sci U S A. 2004;101(19):7347-7351
19. Fritsch L. et al. Conditional gene knock-down by CRE-dependent short interfering RNAs. EMBO Rep. 2004;5(2):178-182
20. Clark IE. et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441(7097):1162-1166
21. Park J. et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441(7097):1157-1161
22. Xia XG. et al. An RNAi strategy for treatment of amyotrophic lateral sclerosis caused by mutant Cu,Zn superoxide dismutase. J Neurochem. 2005;92(2):362-367
23. Lee G, Saito I. Role of nucleotide sequences of loxP spacer region in Cre-mediated recombination. Gene. 1998;216(1):55-65
24. Xia XG. et al. An enhanced U6 promoter for synthesis of short hairpin RNA. Nucl Acids Res. 2003;31(17):e100
25. Tummala H. et al. Inhibition of chaperone activity is a shared property of several Cu,Zn-superoxide dismutase mutants that cause amyotrophic lateral sclerosis. J Biol Chem. 2005;280(18):17725-17731
26. Xia XG. et al. Gene transfer of the JNK interacting protein-1 protects dopaminergic neurons in the MPTP model of Parkinson's disease. Proc Natl Acad Sci U S A. 2001;98(18):10433-10438
27. Xia XG, Harding T, Weller M, Bieneman A, Uney JB, Schulz JB. Gene transfer of the JNK interacting protein-1 protects dopaminergic neurons in the MPTP model of Parkinson's disease. Proc Natl Acad Sci U S A. 2001;98(18):10433-10438
28. Schwarz DS. et al. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115(2):199-208
29. Hornung V. et al. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med. 2005;11(3):263-270
30. Bridge AJ. et al. Induction of an interferon response by RNAi vectors in mammalian cells. Nat Genet. 2003;34(3):263-264
31. Grimm D. et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441(7092):537-541
32. Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23(24):5080-5081
33. Yu J. et al. Transgenic RNAi-mediated reduction of MSY2 in mouse oocytes results in reduced fertility. Dev Biol. 2004;268(1):195-206
34. Golding MC. et al. Suppression of prion protein in livestock by RNA interference. Proc Natl Acad Sci U S A. 2006;103(14):5285-5290
35. Hasuwa H, Kaseda K, Einarsdottir T, Okabe M. Small interfering RNA and gene silencing in transgenic mice and rats. FEBS Lett. 2002;532(1-2):227-230
36. Fedoriw AM. et al. Transgenic RNAi reveals essential function for CTCF in H19 gene imprinting. Science. 2004;303(5655):238-240
37. Chang HS, Lin CH, Chen YC, Yu WC. Using siRNA technique to generate transgenic animals with spatiotemporal and conditional gene knockdown. Am J Pathol. 2004;165(5):1535-1541
38. Stunkel W, Kober I, Seifart KH. A nucleosome positioned in the distal promoter region activates transcription of the human U6 gene. Mol Cell Biol. 1997;17(8):4397-4405
39. Jensen RC, Wang Y, Hardin SB, Stumph WE. The proximal sequence element (PSE) plays a major role in establishing the RNA polymerase specificity of Drosophila U-snRNA genes. Nucleic Acids Res. 1998;26(2):616-622
40. Bonifati V. et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299(5604):256-259
41. Kitada T. et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605-608
42. Valente EM. et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304(5674):1158-1160
43. Itier JM. et al. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet. 2003;12(18):2277-2291
44. Goldberg MS. et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278(44):43628-43635
45. Von Coelln R. et al. Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc Natl Acad Sci U S A. 2004;101(29):10744-10749
46. Perez FA, Palmiter RD. Parkin-deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci U S A. 2005;102(6):2174-2179
47. Chen L. et al. Age-dependent motor deficits and dopaminergic dysfunction in DJ-1 null mice. J Biol Chem. 2005;280(22):21418-21426
48. Goldberg MS. et al. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron. 2005;45(4):489-496
49. Kim RH. et al. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc Natl Acad Sci U S A. 2005;102(14):5215-5220
50. Green JC. et al. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100(7):4078-4083
Author contact
![]() Correspondence to: Xu Gang Xia, Department of Pathology, Anatomy & Cell Biology, Thomas Jefferson University, 508 JAH, 1020 Locust Avenue, Philadelphia, PA 19107, USA. Phone: 215-503-9152; Fax: 215-923-3808; E-mail: xugang.xiaedu. Zuoshang Xu, Department of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School, 364 Plantation St, Worcester, MA 01605.
Correspondence to: Xu Gang Xia, Department of Pathology, Anatomy & Cell Biology, Thomas Jefferson University, 508 JAH, 1020 Locust Avenue, Philadelphia, PA 19107, USA. Phone: 215-503-9152; Fax: 215-923-3808; E-mail: xugang.xiaedu. Zuoshang Xu, Department of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School, 364 Plantation St, Worcester, MA 01605.