Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(5):463-471. doi:10.7150/ijbs.5404 This issue Cite
Research Paper
Karyotypic and Molecular Genetic Changes Associated With Fetal Cardiovascular Abnormalities: Results of a Retrospective 4-Year Ultrasonic Diagnosis Study
Bihui Bao1,2,*, Yu Wang2,*, Hua Hu1, Hong Yao1, Yuyan Li1, Shuai Tang1, Lihong Zheng2, Yan Xu1, Zhiqing Liang1, ![]()
1. Department of Gynecology and Obstetrics, Southwest Hospital, Third Military Medical University, Chongqing 400038, China;
2. Department of Gynecology and Obstetrics, Chengdu Military General Hospital, Chengdu 610083, China.
* These authors equally contributed to this study.
Received 2012-10-17; Accepted 2013-4-22; Published 2013-5-9
Abstract
Objective: To investigate the incidence of aneuploidy in fetuses with congenital heart defects (CHDs) and to further identify submicroscopic changes and global DNA methylation levels as potential biomarkers in complex CHD cases.
Methods: Fetuses at high risk for birth defects or with obvious sonographic anomalies were recruited at the Prenatal Diagnosis Center and Ultrasonic Diagnosis Center. Elective fetal karyotyping and DNA copy number and promoter methylation analyses were carried out following parental consent. G-banded karyotyping was performed to detect fetal aneuploidy. Copy number variations (CNVs) were detected using the Affymetrix SNP Array 6.0 and validated by real time PCR. Global DNA methylation analyses were conducted using a Roche NimbleGen Human DNA Methylation 3x720K Array, and DNA methylation differences were assayed by a Sequenom MassARRAY EpiTYPER.
Results: Conventional karyotyping identified 30 cases with aneuploidy in 179 CHD fetuses. Various CNVs were found in two aneuploid fetuses and in five euploid CHD fetuses. Verified segmental deletion or duplications were not directly associated with cardiovascular malformations except in DAAM1 and GATA6. Verifiable aberrant DNA methylation could not be identified in three complex CHD fetuses.
Conclusions: In this study, Trisomy 18, Trisomy 21 and 45,XO were the most common aneuploidies identified in CHD fetuses. In the affected samples, only DAAM1 deletion and GATA6 amplification could be associated with cardiovascular biological processes.
Keywords: Congenital heart defect, Karyotyping, Copy number variants, Methylation level.
Introduction
Congenital heart defects (CHDs) are the most common birth defects and are the leading cause of infant morbidity and mortality.[1] Despite early surgical palliation of CHDs in neonates with chromosomal aberrations, prognosis can be worse due to the impact of other genetic anomalies. [2, 3] Advances in ultrasound resolution have helped detect fetal structural malformations earlier in gestation.
Many etiological factors, including embryonic chromosomal abnormality, gene mutation, maternal age, disease, malnutrition, hypoxia, radiation, microbial infection and chemical agents, are closely associated with CHD.[1, 4] However, even though syndromic CHDs have similar genetic etiologies, the genetic basis and fundamental etiology for the majority of CHDs remains largely unknown.[5] Karyotyping is typically used in individuals with congenital malformations to identify chromosome defects, including aneuploidy, polyploidy, deletion, duplication, inversions and translocations. Genome-wide copy number analysis can be used to identify chromosomal submicroscopic deletions or duplications in a range of target genetic loci. In addition to primary genetic factors, epigenetic modification may play an important role in CHD causation. For example, folate deficiency, which is associated with fetal CHD and Down syndrome [6] and is a risk factor for developmental abnormalities, is related to the maternal DNA hypomethylation status.
Recurrent chromosomal microdeletions or microduplications can cause CNVs that affect genes involved in cardiac development.[7] CNVs at 1q21.1 and deletions at 15q11.2 or implicating Wnt signaling have been strongly associated with the risk of sporadic, nonsyndromic CHD. [8] The epigenetic status of genes or repeat sequences has a profound effect on cell physiology and long-term developmental effects of the fetus. Thus, changes in DNA methylation may have serious consequences for embryonic and fetal development.[9] In this study, we investigated the incidence of chromosomal abnormalities in CHD fetuses to identify regions harboring disease-related genes and epigenetic changes that may contribute to CHD pathophysiology. We performed DNA CNV and methylation analyses on samples from fetuses with complex CHD to determine biomarkers that may be useful for early prenatal diagnosis. Finally, since many fetuses with aneuploidy have normal cardiac development and some fetuses with complex CHDs have extra-cardiac malformations and a normal karyotype, we also evaluated potential environmental risk factors for adverse fetal effects.
Methods
Ethical approval and recruitment protocol
All clinical research protocols were approved by the Hospital Ethics Committee in Chongqing. After identifying a fetus with CHD, the parents were approached to request consent for records and genetic testing. Signed informed consent was essential before any fetus was entered into the study. Detailed information from the original medical records and reports was collected from the Prenatal Diagnosis Center and the Ultrasonic Diagnosis Center at the Southwest Hospital in Chongqing, China.
Routine prenatal screening for fetal defects
Antenatal care in China includes active prenatal screening in the late first and early second trimesters to detect severe birth defects. Amniocentesis was offered at 19-23 weeks and cordocentesis at 24-40 weeks gestation for fetal karyotyping and DNA testing if the fetal nuchal translucency measurement was ≥ 2.5 mm at 11-13 weeks gestation following an ultrasonic scan, the maternal reproductive age was ≥ 35 years, or the maternal serum triple test [Alpha-fetoprotein (AFP), human chorionic gonadotrophin beta subunit (β-hCG) and unconjugated Estriol (u-E3)] at 15-20 weeks gestation indicated an elevated risk of Trisomy 21 (T21; risk threshold ≥ 1:250), Trisomy 18 (T18; risk threshold ≥ 1:350) or a neural tube defect (AFP MoM ≥ 2.5). In addition, a detailed ultrasound scan was performed at 22-28 weeks gestation, with particular attention to cephalofacial, limb and major visceral organ development. Any structural anomalies diagnosed by ultrasound scanning were monitored and investigated by the registry staff. Definitive CHDs were diagnosed by echocardiography or fetal cardiovascular magnetic resonance imaging (MRI) (repeated 4-6 weeks later if necessary).
Investigating pathogenic factors for CHD
To investigate correlations between family history and CHDs, a detailed questionnaire was given to the parents. The questionnaire requested information regarding any physical or mental illnesses affecting the parents, as well as information regarding any known teratogenic exposure or occupational or leisure time exposure to drugs and environmental chemicals known to cause congenital malformations, such as steroids, antipsychotics, antihistamines, diuretics, cancer chemotherapeutic agents, insect repellents and others. Data on maternal or paternal smoking, drinking, vitamin use, and age were also collected.
Microscopic and submicroscopic chromosomal imbalance studies
With a signed informed consent, fetal cells were sampled from amniotic fluid via amniocentesis and/or cord blood by cordocentesis before legal termination of the pregnancy using mifepristone and misoprostol or in combination with rivano.
Using the Promega Wizard DNA extraction kit (Promega Corp., Madison, WI, USA), DNA from fetuses with severe cardiac anomalies or a single cardiovascular malformation with or without extracardiac malformations was obtained from umbilical cord blood and/or frozen myocardial tissue collected during autopsy after elective pregnancy termination. Samples of parental peripheral blood were also collected for genetic analysis. Other control samples were collected from umbilical cord blood of unrelated normal neonates, fetuses terminated for nonmedical reasons, and peripheral blood from healthy volunteers. Samples were processed immediately or stored at -20 °C for a maximum of 1 month or at -80 °C for longer periods to ensure sample quality.
G-banded karyotyping was performed to detect gross genetic changes at the Prenatal Diagnosis Center. Submicroscopic genetic variations were detected using the Affymetrix SNP Array 6.0 at the National Engineering Research Center for Beijing Biochip Technology in Beijing, China. DNA copy numbers less than 1.5 were regarded as deletions, while DNA copy numbers more than 2.5 were considered as amplifications. DNA was extracted from umbilical cord blood and frozen myocardial tissue and compared to whole blood from the parents, healthy volunteers and unrelated neonates. Significant copy number amplifications and deletions were validated by quantitative real time polymerase chain reaction (qPCR) using a Roche Lightcycler™ automatic quantitative PCR analyzer at the National Engineering Research Center for Beijing Biochip Technology. [10] All validated CNVs were queried in the Database of Genomic Variants (http://projects.tcag.ca/variation/) and the UCSC Genome Browser (http://genome.ucsc.edu/cgi-bin/hgGateway). Information regarding genes affected by the CNVs was identified using the Human Genome Browser (NCBI36/hg18) [11].
Epigenetic mechanism studies
A global methylation assay of CHD fetuses was carried out using a Roche NimbleGen Human DNA Methylation 3x720K CpG Island Plus RefSeq Promoter Array Kit at the National Engineering Research Center for Beijing Biochip Technology. The CHD fetuses were compared with normal fetuses (controls) matched for gender, gestational age at birth and maternal age. The 3x720K microarrays focused on biologically significant genomic regions, including 27,728 CpG Islands and 22,532 RefSeq gene promoters (UCSC, hg18), for unbiased discovery of methylated DNA regions. Global DNA methylation levels between CHD fetuses and controls were measured simultaneously with the same NimbleGen Array kit. DNA methylation changes between CHD cases and controls were assayed again using the same cord blood DNA with a Sequenom MassARRAY EpiTYPER at the National Engineering Research Center for Beijing Biochip Technology to further validate and assess the reliability and validity of their pathogenic potential.
Genetic Testing Quality Assurance
All the samples were analyzed within 1 month of collection, and thus genetic testing was carried out over a 4-year period. To ensure that the assays functioned correctly, the following quality assurance systems were used.
All laboratories participated in external quality assurance schemes for genetic testing. When it was necessary to transfer samples between laboratories, specimens were transported by air with dry ice.
Stored DNA from previous studies was analyzed to ensure similar results. After UV spectrophotometry for the quantitative analysis of nucleic acids and agarose gel electrophoresis of DNA, an aliquot of amplified DNA was stored at -80 °C for use as a control for the next experiment.
In addition, internal quality control samples were used. Before a DNA microarray experiment, quality control samples were used to test the UV spectrophotometry and gel electrophoresis systems.
Statistical analysis
Results were expressed as mean ± standard error of the mean for parametric data. The rate of chromosomal imbalances and risk factors in pregnancy were estimated by respective percentage. CNVs were determined with qPCR probe signals (in log2 ratios) using the hierarchical mode analysis (HMA) algorithm [11]. Real-time qPCR verification of the microarray CNVs was carried out using unpaired students t-tests. [10] Genome-wide DNA methylation analyses in CHD fetuses were based on a case-control study design. P<0.05 was considered to be significant.
Results
Karyotyping of fetuses with heart defects
In total, 1,660 birth defect cases, including 537 CHD cases, were diagnosed prenatally during January 2008 to December 2011 in the Southwest hospital. Of the 537 CHD cases, 33.3% (179/537) were conventionally karyotyped, of which the majority (83.2%, 149/179) had a normal karyotype. Only 16.8% (30/179) of the karyotyped cases showed aneuploidy, including 9 fetuses with T21, 14 fetuses with T18, 1 with Trisomy 13 (T13), 5 with monosomy X (45,XO) and 1 with 69,XXX. T21, T18 and 45,XO were more frequent among aneuploidy fetuses with CHD (Table 1).
Frequency of echocardiographic diagnosis of CHD in the 179 fetuses reported by karyotype.
| Heart defects | Normal karyotype (n=149) | Trisomy 18 (n=14) | Trisomy 21 (n=9) | 45, XO (n=5) | Trisomy 13 (n=1) | 69, XXX (n=1) |
|---|---|---|---|---|---|---|
| VSD | 45 | 9 | 7 | 2 | 1 | 0 |
| AVV dysplasia | 34 | 1 | 1 | 1 | 0 | 0 |
| ECD | 21 | 6 | 2 | 0 | 0 | 1 |
| ASD | 17 | 0 | 0 | 0 | 0 | 0 |
| PS | 17 | 0 | 0 | 0 | 0 | 0 |
| PTA | 16 | 1 | 3 | 0 | 0 | 0 |
| Dextrocardia | 15 | 0 | 0 | 0 | 0 | 0 |
| TGA | 14 | 0 | 0 | 0 | 0 | 0 |
| Hyperplastic ventricle | 13 | 1 | 0 | 1 | 0 | 0 |
| TOF | 11 | 4 | 1 | 2 | 0 | 0 |
| Arrhythmia | 1 | 0 | 1 | 1 | 0 | 0 |
| Pericardial effusion | 1 | 0 | 0 | 1 | 0 | 0 |
| AVSD | 8 | 4 | 0 | 1 | 0 | 0 |
| Right AA | 8 | 0 | 0 | 0 | 0 | 0 |
| PLSVC | 6 | 1 | 1 | 0 | 0 | 0 |
| Expansion of CS | 6 | 1 | 1 | 0 | 0 | 0 |
Abbreviations: AA-aortic arch, ASD-atrial septal defect, AVSD-atrioventricular septal heart defect, AVV dysplasia -atrioventricular valve dysplasia (mitral and/or tricuspid stenosis, regurgitation), CS-coronary sinus, ECD-endocardial cushion defect, PS-pulmonary stenosis, PTA-persistent truncus arteriosus, PLSVC-persistent left superior vena cava, TGA-transposition of great arteries, TOF-tetralogy of Fallot, VSD-ventricular septal defect.
During the 4-year period, CHD-related aneuploidy occurred in 96 cases of diverse birth defects, including 30 CHD cases (Table 2). There were 64 prenatally-detected cases of T21 (including 8 mosaics and 2 translocations). The CHD incidence of T21 was therefore 14.1% (9/64). Similarly, there were 19 cases of T18, with a CHD incidence of 73.7% (14/19). 45,XO occurred in 11 fetuses, including a 45,XO(40)/46,XX(11) mosaicism and a structural aberration of 45,XO/46,X,r(X) (71.7%: 28.3%), with a CHD incidence of 45.5% (5/11). Both cases of T13 and 69,XXX showed CHDs. Overall, the incidence of fetal cardiac malformations in CHD-related aneuploidy cases was 31.3% (30/96).
Incidence of cardiac and extra-cardiac malformations in fetuses with CHD-related karyotypes.
| Karyotype (n) | One heart defect, n (%) | >1 heart defect, n (%) | Extracardiac malformations, n (%) |
|---|---|---|---|
| Normal karyotype (149) | 41 (27.5) | 108 (72.5) | 42 (28.2) |
| Trisomy 21 (64) | 3 (4.7) | 6 (9.4) | 64 (100) |
| Trisomy 18 (19) | 4 (21.1) | 10 (52.6) | 19 (100) |
| Monosomy X (11) | 2 (18.2) | 3 (27.3) | 11 (100) |
| Trisomy 13 (1) | 1 | 0 | 1 |
| Triploidy (1) | 0 | 1 | 1 |
Major congenital malformations of CHD fetuses
A list of the heart defects identified in the 179 cases is presented in Table 1. Ventricular or atrioventricular septal defects and endocardial cushion defects (ECD) comprised 71.5% of the heart defects. One cardiovascular malformation occurred in 21.2% (38/179) of the cases. Most cases were complex cardiac malformations (70.9%, 127/179) and/or were associated with extracardiac malformations (40.2%, 72/179). The most common extra-cardiac malformations occurred in the gastrointestinal system, central nervous system and urinary tract. All of the CHD aneuploidy cases (30) had various extra-cardiac malformations. In contrast, only 28.2% (42/149) of the CHD cases with a normal karyotype had extra-cardiac defects. Polyhydramnios or oligohydramnios, single umbilical artery (SUA), or an elevated umbilical artery peak-systolic to end-diastolic blood flow velocities ratio (S/D values) occurred in 70.0% (21/30) of the CHD fetuses with aneuploidy, but occurred in only 8.72% (13/149) of the CHD fetuses with normal karyotypes. Detailed cardiac abnormalities are shown in Table 1 and Table 2.
The aneuploidy most frequently associated with CHDs was T18, which had characteristic large, prominent ventricular septal defects (VSDs, 9/14), ECDs (6/14), atrioventricular septal defects (AVSD) (4/14) and Tetralogy of Fallot (TOF) (4/14). Sonographic markers of T18 were cephalofacial anomalies and anomalies of the extremities, including choroid plexus cysts (n=5), cleft lip and palate (CLP, n=3), low-set ears (n=2), micrognathia (n=2), a short radius and ulna (n=2), and overlapping fingers with clenched fists and club feet (n=1). Polyhydramnios (8/14) and SUA (6/14) were also identified.
The most common aneuploidy was T21 (64 cases). Heart malformations identified in 9 cases included peri-membranous VSD (n=7), persistent truncus arteriosus (PTA) (n=3), ECD (n=2), TOF (n=1) and arrhythmia (n=1). Extra-cardiac malformations were characterized by the absence or hypoplasia of the nasal bone (n=7), gastrointestinal anomalies (n=4), cerebral ventricular dilatation (n=2), micrognathia (n=1), ocular hypertelorism (n=1), an extended tongue (n=1), polyhydramnios (n=6), and elevated umbilical artery S/D values (n=4).
The 45,XO cases (5) were associated with VSD, TOF, AVSD, edema, nuchal cystic hygroma, and oligohydramnio. The case with 69,XXX had complete ECD, intrauterine growth restriction and oligohydramnio. The T13 case had a large VSD (4.1 mm diameter), prominent microphthalmia, and microcephaly.
DNA CNV analyses of CHD fetuses
Seven CHD fetuses with particularly complex anomalies underwent genome-wide CNV analysis, including 5 with a normal karyotype, 1 with T18, and 1 with 45, XO. Besides previously implicated candidate genes related to cardiac development, a diverse range of 50 Kb-1.6 Mb DNA copy number amplifications or deletions were present in the 5 fetuses with a normal karyotype. However, most of the apparently significant CNVs were false positives that could not be verified by real-time qPCR when compared with the parents, 6 healthy volunteers and 8 unrelated neonates. [10] A previously undescribed 286 Kb single-copy deletion at 14q23.1 from 58678231 to 58964009 (NCBI36/hg18), which contained whole DAAM1 and KIAA0666 genes, existed in one normal karyotype fetus, which was diagnosed with a single atrium, pulmonary stenosis, double outlet right ventricle (DORV), dextrocardia, complete ECD and hydropericardium. Although this verified deletion could not with certainty be associated with these heart defects, [10] studies using DAAM1-deficient mice revealed that DAAM1 was essential for cardiac morphogenesis. [12]
In the genome of the T18 fetus, over 60 segments (56 Kb-1.2 Mb) had copy number amplifications and contained protein-coding genes, non-coding RNA (ncRNA) genes, open reading frames, and pseudogenes. The identified copy number amplification genes, such as ZNF521, RNF138, RAB12, ATP5A1, PTPN2 and CTAGE1, are involved in multiple cellular processes, including transcription, proliferation, differentiation, migration and immunity. On chromosome 18 (Hsa18), GATA6 was the only critical gene involved in cardiac differentiation and cardiomyocyte proliferation. Triploid GATA6 might have been significantly related to the high incidence of CHDs in T18 cases [13].
In the 45,XO fetal genome, there were approximately 45 discrete regions on the X chromosome with a single copy number, including 7 segments from 243 Kb to 1.7 Mb. These copy number deletion regions contained various protein-coding genes, ncRNAs and pseudogenes involving system, anatomical structure and organismal development. However, the identified CNVs that spanned genes on the X chromosome were not clearly involved in cardiovascular biological processes.
DNA methylation detection in CHD fetuses
Umbilical cord blood samples from three fetuses with a single atrioventricular valve, ECD and transposition of great vessels were analysed for their DNA methylation state. Three normal fetuses were matched by gender (2 male and 1 female), gestational age (24-28 weeks) and maternal age (24~27 years) as controls. The Log2-ratio of methylation and unmethylation intensities for quantifying methylation levels increased on average by 0.41-0.59 in three affected cases in the promoter region of RADIL (Figure 1), CHRNA10 and APOB genes (data not shown). In contrast, the ratio was reduced by 0.34-0.62 in the promoter region of NXPH3, ZNF408 and ABL1 in the affected cases (data not shown). The relative hyper- and hypomethylated candidate genes were identified from UCSC human genome annotations (GRCh37/hg19) for their potential biological significance, and methylation regions with dramatic changes were selected for subsequent analyses using a Sequenom MassARRAY system with the same cases and controls. Quantitative methylation analyses in the same promoter region of the above genes within CpG islands did not show noticeable low or high methylation levels, including ZNF408, ABL1, APOB and RADIL (Figure 2). Gene-specific methylation patterns did not deviate significantly as previously observed.
SignalMap software graph of differential DNA methylation in the promoter region of RADIL using a Human Meth 3x720K microarray. Upper green bars match Log2-ratio and region features of the RADIL gene. The differentially methylated region in chromosome 7, 4891350-4892250, was identified with cases (lower three strips on the above 1/3 part) higher than 0.81-1.08 when compared with controls (upper three strips on the above 1/3 part) using NimbleScan software. Lower green horizontal bars demonstrate the methylation peak score that showed significant methylation differences. Middle black, brown and blue horizontal bars indicate CpG islands, primary transcripts and tiled regions, respectively, on the array design. The light blue box highlights the differentially methylated region between the cases and controls.

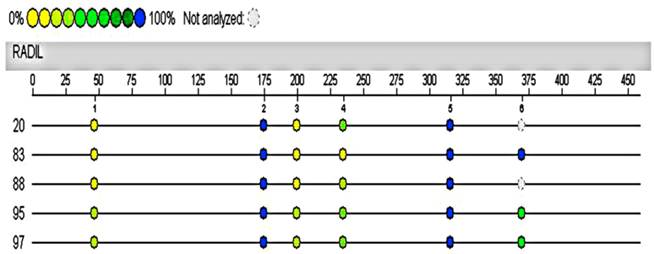
Hypermethylated RADIL region in 4891333-4891902 of chromosome 7 was assayed again with the Sequenom MassARRAY system using the same cases and controls. The automated analysis result indicated no methylation differences in the promoter of RADIL between the cases (No.20, 95, 97) and controls (No.83, 88, the remaining DNA of the other control was not sufficient for the following test).
Potential risk factors for CHDs
The family history data did not indicate a familial origin for CHD in any of the 179 fetuses. However, other factors did indicate significant differences. The mean maternal age of a CHD fetus with aneuploidy was 4.6 years older than that with a normal karyotype. Detailed information of maternal age is presented in Table 3. Environmental factors reported for CHD risk included a history of cold (n=35), fever (n=12), medicine (n=49), recent housing renovation (n=20), living close to high-voltage electricity pylons (n=2), and living close to a leather workshop (n=2) during early gestation.
Maternal age of the CHD fetuses.
| Groups of CHD fetuses | Mean maternal age (years) | Number of mother over 30 years (%) | Number of mother over 35 years (%) |
|---|---|---|---|
| aneuploidy (30) | 32.3±5.8 | 19 (63.3) | 10 (33.3) |
| Normal karyotype (149) | 27.7±4.9 | 39 (26.2) | 12(8.1) |
| All CHD cases (179) | 28.2±5.3 | 58 (32.4) | 22 (12.3) |
Discussion
Prenatal screening for birth defects can be performed starting at the late first trimester (9-11 weeks onwards).[14] Nuchal thickening is used as an index of aneuploidy and is associated with fetal cardiac strain, AVSD or VSD, a narrow aortic isthmus, hypoplastic left heart, and abnormal development of lymphatic vessels and the jugular vein.[1, 3] Aneuploidy causes altered gene dosage and results in developmental abnormalities, including Down, Edwards, Patau and Turner syndromes. [15, 16] CHDs complicated by significant extra-cardiac malformations suggest karyotypic abnormalities. There are multiple loci involved in cardiogenesis, including chromosomes 1, 3, 4, 8, 11 and 22.[17] Therefore, in this study, we investigated the abnormal karyotype incidence in a small group of CHD fetuses. Syndromic CHDs were commonly associated with T18, T21 and 45,XO, but the majority of CHD cases were non-syndromic cardiac malformations. We focused on DNA CNV and global methylation changes for their potential use in genetic counseling and risk assessment.
In this study, there were fewer aneuploidy cases than in other studies. This may be due to the small number of patients that underwent routine karyotyping, a lack of autopsy in most cases, an incomplete penetrance phenotype for some extra-cardiac malformations, or differential referral patterns to specialized centers.[3, 18] Also, with extensive use of second-trimester fetal anomaly scanning, craniofacial defects and abnormal gastrointestinal tracts suggest aneuploidy and many parents now opt for invasive diagnostic testing with prompt karyotyping or specific qPCR before obstetric management.
Trisomies cause a plethora of phenotypic alterations due to extra genetic material affecting cell and tissue homeostasis.[19] Overexpression of COL6A1 and COL6A2 in T21 induces collagen type VI formation along the epidermal basement membrane of the subcutis, causing increased nuchal translucency.[20] In our study, CNV analyses showed that some genes and ncRNA had a 50% copy number amplification in T18, a 50% decrease in 45,XO, and various CNVs in cases with normal karyotypes. However, only DAAM1 was associated with cardiac morphogenesis. [21] Interestingly, this finding was consistent with previous analyses in DAAM1-mutant mice, which had DORV, VSD and pericardial effusion. These similar cardiac defects were probably caused by abnormal DAAM1-mediated actin cytoskeletal regulation and cardiogenesis.[12] Thus, copy number deleted regions on 14q23.1 may be implicated in the etiology of complex CHDs.
Human heart development requires an orderly coordination of transcriptional programs with the transcription factors GATA4, GATA6, Tbx5, NKX2-5, and others. Lack of NKX2-5 in animal models has been shown to cause degeneration of cardiac myocytes, impairment of cardiac looping and embryonic lethality.[22] Nearly 100% of mice heterozygous for GATA4 and Tbx5 gene defects suffered embryonic or neonatal lethality. Common defects in these animals included complete AVSDs with a single AV valve and myocardial thinning.[13] Mutations in GATA4 and NKX2-5 resulted in transcriptional inactivation and were subsequently responsible for VSD, TOF, ECD, hypoplastic left heart syndrome, DORV, L-/D-transposition of the great arteries, tricuspid valve abnormalities, and conduction anomalies.[23] However, these causal links are only able to explain a very small fraction of human CHDs. Over-expression of Hsa18 genes interact with GATA4, Tbx5, NKX2-5 or MYH6, may disrupt cardiovascular development, and increase the risk of CHD in Edwards' syndrome.[13] Moreover, defects in GATA6 increase the incidence of CHD.[19] However, not all T18 exhibit syndromic CHDs due to Hsa18 gene duplication. In addition, fetuses with a 45,XO karyotype and unbalanced X:A expression ratios are known as nuchal cystic hygroma due to jugular lymphatic obstruction. Consequently, hemodynamic disturbances and heart valve regurgitation secondarily perturb cardiac morphogenesis, resulting in hypoplastic great vessels and partial heart hypertrophy. In this study, a potential X chromosome region was not associated with human cardiovascular morphogenesis in our CHD cases.
Endothelial cells play a crucial role in the formation of the endocardial cushion and AV valves and septa. However, the transformation mechanisms of endothelial cells to mature mitral and tricuspid valves and AV septa are not well understood.[25, 26] Perturbation signals, including variant genes and adverse environmental triggers, may disrupt proliferation and differentiation of endocardial cells, causing AV septal and valve defects.[25] Nutritional deficiencies, tobacco and alcohol consumption, toxoplasma or viral infections, illness, radiation and toxic chemicals may play a crucial role in CHD development.[17, 22, 27, 28] The incidence of CHDs can be decreased by 25-50% with folic acid supplements due to the protective effects of folic acid on embryonic and fetal cardiomyocytes against environmental toxins and parental diseases.[17]
The methylation status of CpG islands in the promoter region of genes has a profound effect on cell physiology, histological differentiation and embryonic development. During embryogenesis and the early development of the fetus, DNA methylation undergoes dramatic reprogramming and varies depending on cell type.[29] Inefficient epigenetic reprogramming can decrease cell proliferation and impair embryonic development.[9] Aberrant DNA methylation patterns can lead to birth malformations and human carcinoma.[30] In this study, we determined whether CHD fetuses suffered CHD-specific methylation pattern disturbance. The results showed no drastic changes in the DNA methylation patterns, but there were alterations of methylation levels in the promoter regions of several genes, such as the hypermethylation of RADIL and hypomethylation of ZNF408. RADIL is required for cell adhesion and migration of neural crest precursors during development, while ZNF408 is related with heart development (GRCh37/hg19). Using the Sequenom MassARRAY system, no significant methylation differences could be demonstrated in the promoter regions between the CHD cases and controls. A larger study is therefore advisable in the future to clarify epigenetic mechanisms in CHD pathophysiology.
Although elucidating the molecular basis of a given heart defect is challenging, complex trait analyses and causal investigation of CHDs can further our understanding and lead to better genetic counselling of patients with CHDs. The maternal serum “triple marker screen” and ultrasound scans are available as prenatal noninvasive screening tests of some serious congenital anomalies. Invasive diagnostic tests, including chorionic villus sampling, amniocentesis and cordocentesis, are performed for fetal karyotyping, DNA analysis and biochemical testing to help further understand the genetic basis of some CHDs.
Acknowledgements
We acknowledge the participation of families and thank Professors Qing Chang, Jing Cheng and Yimin Sun for sample collection and their excellent technical assistance.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tennstedt C, Chaoui R, Korner H, Dietel M. Spectrum of congenital heart defects and extracardiac malformations associated with chromosomal abnormalities: results of a seven year necropsy study. Heart. 1999;82:34-9
2. Poprawski K, Michalski M, Lawniczak M, Lacka K. Cardiovascular abnormalities in patients with Turner syndrome according to karyotype: own experience and literature review. Pol Arch Med Wewn. 2009;119:453-60
3. Wimalasundera RC, Gardiner HM. Congenital heart disease and aneuploidy. Prenat Diagn. 2004;24:1116-22
4. Hameed AB, Sklansky MS. Pregnancy: maternal and fetal heart disease. Curr Probl Cardiol. 2007;32:419-94
5. Priest JR, Girirajan S, Vu TH, Olson A, Eichler EE, Portman MA. Rare copy number variants in isolated sporadic and syndromic atrioventricular septal defects. Am J Med Genet A. 2012;158A(6):1279-1284
6. van Driel LM, de Jonge R, Helbing WA, van Zelst BD, Ottenkamp J, Steegers EA, Steegers-Theunissen RP. Maternal global methylation status and risk of congenital heart diseases. Obstet Gynecol. 2008;112:277-83
7. Goldmuntz E, Paluru P, Glessner J. et al. Microdeletions and microduplications in patients with congenital heart disease and multiple congenital anomalies. Congenit Heart Dis. 2011;6(6):592-602
8. Soemedi R, Wilson IJ, Bentham J. et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am J Hum Genet. 2012;91(3):489-501
9. Yin LJ, Zhang Y, Lv PP. et al. Insufficient maintenance DNA methylation is associated with abnormal embryonic development. BMC Med. 2012;10:26
10. Bao B, Zhang L, Hu H, Yin S, Liang Z. Deletion of a single-copy DAAM1 gene in congenital heart defect: a case report. BMC Med Genet. 2012;13:63
11. Affymetrix. Affymetrix® genome-wide human SNP Nsp/Sty 6.0 user guide; P/N 702504, Rev 3. Affymetrix Inc. 2008:28-332.
12. Li D, Hallett MA, Zhu W. et al. Dishevelled-associated activator of morphogenesis 1 (Daam1) is required for heart morphogenesis. Development. 2011;138(2):303-315
13. Maitra M, Schluterman MK, Nichols HA. et al. Interaction of Gata4 and Gata6 with Tbx5 is critical for normal cardiac development. Dev Biol. 2009;326:368-77
14. Sieroszewski P, Perenc M, Bas-Budecka E, Suzin J. Ultrasound diagnostic schema for the determination of increased risk for chromosomal fetal aneuploidies in the first half of pregnancy. J Appl Genet. 2006;47:177-85
15. Hsiao CC, Tsao LY, Chen HN, Chiu HY, Chang WC. Changing clinical presentations and survival pattern in trisomy 18. Pediatr Neonatol. 2009;50:147-51
16. Iliopoulos D, Vassiliou G, Sekerli E. et al. Long survival in a 69,XXX triploid infant in Greece. Genet Mol Res. 2005;4:755-9
17. Lin AE, Ardinger H. Genetic epidemiology of cardiovascular malformations. Progr Pediatr Cardiol. 2005;20:113-26
18. Bahado-Singh RO, Choi SJ, Oz U, Mendilcioglu I, Rowther M, Persutte W. Early second-trimester individualized estimation of trisomy 18 risk by ultrasound. Obstet Gynecol. 2003;101:463-8
19. Chen CP. Prenatal sonographic features of fetuses in trisomy 13 pregnancies (I). Taiwan J Obstet Gynecol. 2009;48:210-7
20. von Kaisenberg CS, Brand-Saberi B, Christ B, Vallian S, Farzaneh F, Nicolaides KH. Collagen type VI gene expression in the skin of trisomy 21 fetuses. Obstet Gynecol. 1998;91:319-23
21. Greenway SC, Pereira AC, Lin JC. et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet. 2009;41:931-5
22. Brewer AC, Alexandrovich A, Mjaatvedt CH, Shah AM, Patient RK, Pizzey JA. GATA Factors Lie Upstream of Nkx 2.5 in the Transcriptional Regulatory Cascade That Effects Cardiogenesis. STEM CELLS AND DEVELOPMENT. 2005;14:425-439
23. Ikeda Y, Hiroi Y, Hosoda T. et al. Novel Point Mutation in the Cardiac Transcription Factor CSX/NKX2.5 Associated With Congenital Heart Disease. Circulation Journa. 2002;66:561-563
24. Reamon-Buettner SM, Hecker H, Spanel-Borowski K, Craatz S, Kuenzel E, Borlak J. Novel NKX2-5 Mutations in Diseased Heart Tissues of Patients with Cardiac Malformations. American Journal of Pathology. 2004;164(6):2117-2125
25. Haramis APG, Clevers HC. Holehearted: genetic approaches to congenital cardiac valve malformations. Drug Discovery Today: Disease Mechanisms / Cardiovascular diseases. 2004;1:1-8
26. Nath AK, Krauthammer M, Li P. et al. Proteomic-based detection of a protein cluster dysregulated during cardiovascular development identifies biomarkers of congenital heart defects. PLoS One. 2009;4:e4221
27. Chen BY, Hwang BF, Guo YL. Epidemiology of congenital anomalies in a population-based birth registry in Taiwan, 2002. J Formos Med Assoc. 2009;108:460-8
28. Memon S, Pratten MK. Developmental toxicity of ethanol in chick heart in ovo and in micromass culture can be prevented by addition of vitamin C and folic acid. Reprod Toxicol. 2009;28:262-9
29. Chowdhury S, Erickson SW, MacLeod SL. et al. Maternal genome-wide DNA methylation patterns and congenital heart defects. PLoS One. 2011;6:e16506
30. Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433-40
Author contact
![]() Corresponding author: Zhiqing Liang, Department of Gynecology and Obstetrics, Southwest Hospital, Third Military Medical University, Chongqing 400038, China. Tel: +86-23-68754409; Fax: 86-23-65461867 E-mail: zhi.lzliangcom.
Corresponding author: Zhiqing Liang, Department of Gynecology and Obstetrics, Southwest Hospital, Third Military Medical University, Chongqing 400038, China. Tel: +86-23-68754409; Fax: 86-23-65461867 E-mail: zhi.lzliangcom.