Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(1):59-66. doi:10.7150/ijbs.10174 This issue Cite
Research Paper
Coenzyme Q10 Protects Astrocytes from ROS-Induced Damage through Inhibition of Mitochondria-Mediated Cell Death Pathway
Li Jing1*, Mao-Tao He1*, Yue Chang1, Suresh L. Mehta2,3, Qing-Ping He2, Jian-Zhong Zhang1 ![]() , P. Andy Li2
, P. Andy Li2 ![]()
1. Department of Pathology, Ningxia Medical University and Ningxia Key Laboratory for Cerebrocranial Diseases, Yinchuan, Ningxia, P. R. China.
2. Department of Pharmaceutical Sciences, Biomanufacturing Research Institute and Technological Enterprise (BRITE), North Carolina Central University, Durham, North Carolina, USA.
3. Department of Neurological Surgery, University of Wisconsin, School of Medicine and Public Health, Madison, Wisconsin, USA.
*Co-first authors.
Received 2014-7-23; Accepted 2014-10-14; Published 2015-1-1
Abstract

Coenzyme Q10 (CoQ10) acts by scavenging reactive oxygen species to protect neuronal cells against oxidative stress in neurodegenerative diseases. The present study was designed to examine whether CoQ10 was capable of protecting astrocytes from reactive oxygen species (ROS) mediated damage. For this purpose, ultraviolet B (UVB) irradiation was used as a tool to induce ROS stress to cultured astrocytes. The cells were treated with 10 and 25 μg/ml of CoQ10 for 3 or 24 h prior to the cells being exposed to UVB irradiation and maintained for 24 h post UVB exposure. Cell viability was assessed by MTT conversion assay. Mitochondrial respiration was assessed by respirometer. While superoxide production and mitochondrial membrane potential were measured using fluorescent probes, levels of cytochrome C (cyto-c), cleaved caspase-9, and caspase-8 were detected using Western blotting and/or immunocytochemistry. The results showed that UVB irradiation decreased cell viability and this damaging effect was associated with superoxide accumulation, mitochondrial membrane potential hyperpolarization, mitochondrial respiration suppression, cyto-c release, and the activation of both caspase-9 and -8. Treatment with CoQ10 at two different concentrations started 24 h before UVB exposure significantly increased the cell viability. The protective effect of CoQ10 was associated with reduction in superoxide, normalization of mitochondrial membrane potential, improvement of mitochondrial respiration, inhibition of cyto-c release, suppression of caspase-9. Furthermore, CoQ10 enhanced mitochondrial biogenesis. It is concluded that CoQ10 may protect astrocytes through suppression of oxidative stress, prevention of mitochondrial dysfunction, blockade of mitochondria-mediated cell death pathway, and enhancement of mitochondrial biogenesis.
Keywords: astrocyte, caspase, coenzyme Q10, mitochondrion, reactive oxygen species, ultraviolet.
Introduction
Astroglial cells are located in the brain and optic nerve head [1, 2]. They provide structural, trophic, and metabolic support to neurons and modulating synaptic activity [2-7]. The survival of astroglial cells is critical for repair and recovery of ischemia damaged brain tissue [8-10]. UVB irradiation is known to cause DNA damage and increase reactive oxygen species (ROS). Increased ROS accumulation, either caused by enhancement of production or suppression of anti-oxidant capacity, is highly toxic to many types of cells including astrocytes [11, 12]. The ROS causes lipid peroxidation, protein oxidation and mutagenesis [13]. Inhibition of ROS production can protect cells against oxidative stress-mediated neuronal cell death [14].
Coenzyme Q10 (CoQ10) is an essential cofactor of the electron transport chain. CoQ10 has been shown to protect neuronal cells from UVB and ROS induced damage and to improve neurological behavior in animal models of Parkinson's disease [15]. Either intraocular or oral administration of CoQ10 has been shown to minimize the glutamate toxicity and to protect cells from brain ischemia/reperfusion induced damage in rats [16]. Our previous studies also suggest that CoQ10 could protect neural cells from UVB damage [17]. However, it is not until recently the effects of CoQ10 were studied in astrocytes. The aim of this study is to examine the protective effects of coenzyme CoQ10 on astrocyte cell death and to explore the mechanisms of protection.
Material and Methods
Materials
Dulbecco's modified Eagle's medium (DMEM), phosphate buffered saline (PBS), Fetal bovine serum (FBS), Trypsin-Versene mixture, l-glutamine and penicillin-streptomycin solution were purchased from HyClone laboratories (USA). CoQ10, was purchased from Sigma-Aldrich (USA), dissolved in DMSO (Mediatech, USA) and diluted in culture medium.
Cell Culture, UVB Irradiation and CoQ10 Treatment
Murine astrocytes obtained from embryo (G355-5, ATCC, USA) were cultured in DMEM containing 10% FBS, 2 mM glutamine, and 200 mM streptomycin/penicillin and then maintained at 90%-95% relative humidity in 5% CO2 at 37 °C. The culture media were renewed every 3 days. Cells (15,000-80,000/well) were seeded in 12-well or 96-well cell culture plates and incubated in the above medium for at least 24 h in CO2 incubator to allow 80% confluence. Prior to UVB irradiation, the cultures were washed twice with PBS to remove residual serum and nonattached cells. Cells were incubated for 1 h in serum-free medium and then exposed to 7 mJ/cm2 dose of UVB radiation with a Fisher UV Transilluminator FB-TI-88A. The UVB dose energy is calculated by multiplying the UV intensity by the time of exposure. This dosage was chosen based on our previous study showing 50-70% cell death at 24 h post-exposure. After UVB radiation, cells were returned to the culture incubator for 24 h of recovery at 37°C. Three CoQ10 treatment protocols (each with concentrations of 10 and 25 μg/ml) were used: 1) CoQ10 was given 24 h prior to UVB and maintained during UVB, 2) CoQ10 was treated 24 h prior to UVB and maintained during UVB and 24 h post-UVB, 3) CoQ10 was given 3 h prior to UVB and maintained during UVB and 24 h post-UVB. DMSO (0.1%) treated cells were used as control.
Cell Viability Assay
At 24 h after termination of UVB irradiation, the number of viable cells in 12-well plates was counted using Beckman Coulter Vi-Cell Automated Cell Viability Analyzer (WS-VICELL), which uses the trypan blue exclusion principle. The percent of cell viability was calculated and presented. Cell viabilities were further verified using the MTT (3-(4, 5,-dimethythiazol-2-yl)-2, 5-diphenyl tetrazolium bromide) conversion assay, which also reflects the mitochondrial succinate dehydrogenase activity. Absorbency at the wavelength 570 nm was read on an ELISA plate reader (Molecular Device, San Jose, USA). The results were expressed as a percentage of absorbency relative to control values. All experiments were performed in triplicate and repeated in at least three separate experiments.
Measurements of mitochondrial respiration
The cells were homogenized in 10 volumes of the 1x Extraction Buffer (MITOSIO1, Sigma) containing isotonic solution of mM HEPES (pH 7.5), 200 mM mannitol, 70 mM sucrose, 1 mM EGTA and 2 mg/ml dilapidated BSA. The homogenate was centrifuged at 600 g for 5 min and the supernatant was removed. The pellet was resuspended with 1x extraction buffer and centrifuged at 11,000 g for 10 min. The final pellet was resuspended in 1x extraction buffer (~40 ml/100 mg tissue). Protein concentrations were determined using a microplate BCA protein assay kit (Thermo Scientific) and 0.2 mg of protein was used for respiration studies. Mitochondrial respiration was assessed in a peltier temperature-controlled two chamber high-resolution respirometer (O2K, Oroboros Instruments, Austria) at 37ºC.
Measurements of superoxide and mitochondrial membrane potential
Intracellular ROS (superoxide anion) production was measured using dihydroethidine (DHE) probe in 4 experimental groups. Briefly, cells (2 × 106/mL) were incubated with the DHE (2.5 μM) for 30 min at 37 °C. Cells were washed, resuspended in PBS and analyzed for fluorescence intensity using a Fluoromax-4 spectrofluorometer (HORIBA Jobin Yvon Inc, Edison, NJ) at the excitation and emission wavelengths of 480 nm and 590 nm, respectively. The fluorescence was recorded and presented as relative fluorescence intensity (RFI). Mitochondrial membrane potential was measured using the tetramethylrhodamine methyl ester (TMRM). Briefly, cells (1×106/mL) were harvested and incubated with 30 nM TMRM at 37 °C for 1 h. The cells were washed in PBS and fluorescence measurement was obtained using the Fluoromax-4 spectrofluorometer at excitation and emission of 530 and 573 nm respectively. Mitochondrial potential was dissipated with carbonylcyanide p-trifluoromethoxyphenylhydrazone (FCCP 5 µM) and used as a positive control.
Immunocytochemistry
Astrocytes were fixed in 4% paraformaldehyde for 20 min at room temperature and processed for immunocytochemistry. After permeabilization with 0.1% Trinton X-100, the cells were incubated overnight at 4°C with monoclonal anti-cytochrome C (1:200 dilution, Abcam), polyclonal anti-Bax antibody (1:50 dilution, Abcam) and polyclonal anti-cleaved caspase-9 antibody (1:50 dilution, Cell Signaling Technology) followed by incubation with a secondary donkey anti-goat Alexa Fluor 488 (Invitrogen, Carlsbad, CA, USA) or donkey anti-rabbit Alexa Fluor 567 conjugate (Invitrogen) for 1 h at room temperature. The specimens were mounted with Vectashield Hardset Mounting Media (H-1200) containing DAPI and examined using a fluorescence confocal microscope (Nikon Eclipse C1). Three microscopic fields at 400X were captured and number of positively stained cells was counted.
Western Blot Analysis
After 24 h UVB treatment, cells were collected and lysed on ice in lysis buffer containing 20 mM Tris (pH7.4), 10 mM KCl, 3 mM MgCl2, 0.5% NP40 and complete protease inhibitors (Millipore). The lysates were centrifuged at 500 g for 10 min. The supernatant (S1) fraction was then centrifuged at 20,000 g for 20 min and the resulting supernatant was used as a cytosolic fraction. The pellet (P1) fraction was washed twice with lysis buffer and resuspended in lysis buffer containing 1%SDS. The resulting lysates were sonicated briefly (Misonix, Ultrasonic Cell Disrupter) and then centrifuged at 20,800 g for 30 min. The resulting supernatants were designated as nuclear fractions. The purity of different cellular fractions has been tested previously [18]. Protein contents from each sample were measured using Microplate BCA Protein Assay Kit (Thermo Scientific). Equal amounts of protein (20 µg) were loaded into each lane, separated in 10% NuPAGE BT gels (Invitrogen), transferred to a PVDF membrane (Millipore), and probed with antibodies against cytochrome c (1:500, Millipore), caspase-9 (1:1000, Cell Signaling), or caspase-8 (1:1000, Cell Signaling).
Mitochondrial biogenesis assay
Mitochondrial biogenesis was analyzed with MitoBiogenesis™ In-Cell ELISA Kit (ab110217, ABCAM) according to the manufacturer's protocol. Briefly, 20,000 cell per well were seeded in 96 well plate. These cells were fixed with 4% paraformaldehyde after adhering to the bottom of the plate. Cells were washed with PBS and followed by the addition of 100 μL of freshly prepared 0.5% acetic acid for 5 minutes to block endogenous alkaline phosphatase activity. Cells were washed again with PBS and permeabilized with 0.1% Triton X-100 for 30 min and followed by the addition of 200 μL of 2X Blocking Solution for 2 h. These cells were then incubated with primary antibodies specifically against mtDNA encoded COX-I, and nuclear-DNA encoded SDH-A proteins. Cells were washed with a washing buffer and incubated with AP for SDH-A and HRP for COX-I secondary antibodies. After washing, the reaction was sequentially developed first with AP reagent and then HRP development solution. A 15-min kinetic reaction with 1 min interval was recorded using a microplate reader. Whole cell staining with Janus Green was also done to remove variation due to cell loss during the procedure. The experiment was run in triplicates and at minimum 3 repetition per group. The ratio of COX-I/SDH-A was calculated to determine mitochondrial biogenesis.
Statistical Analysis
Data are presented as means ± S.D. One-way ANOVA followed by Tukey's test was used for statistical analysis. A value of P < 0.05 was considered statistical significant.
Results
UVB Irradiation Induced Astrocyte Death
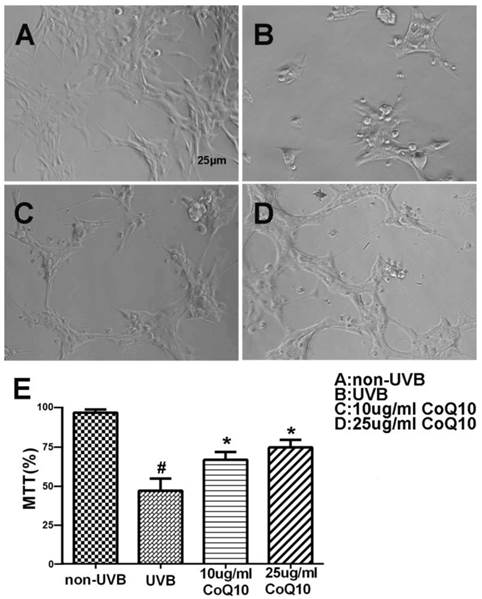
To investigate the effect of CoQ10 on UVB-induced damage to astrocytes, the cell viability was assessed using the MTT assay for non-UVB-challenged (Fig. 1A), UVB-challenged (Fig. 1B), and CoQ10-treated groups (Fig. 1C-D). Addition of 0.1% DMSO did not influence of the cell survival rate after UVB-irradiation. UVB exposure decreased cell viability to 60% of the control value. Treatment with CoQ10 at a concentration of 10 or 25 μg/ml for 24 h prior to UVB exposure and maintaining the same concentration for 24 h after UVB significantly improved cell viability compared with UVB-irradiated astrocytes (Fig. 1E).
Cell viability after CoQ10 treatment in UVB exposed astrocyte. (A-D) Original representative photomicrograms showing cell morphology in different groups; (E) MTT assay showing cell viability in experimental groups. Values are means s.d. from 3 independent experiments. #P < 0.01 vs. non-UVB control and *P < 0.05 vs. UVB. One-way ANOVA followed by Tukey's test. Data are presented as means ±S.D.
Mitochondrial respiration
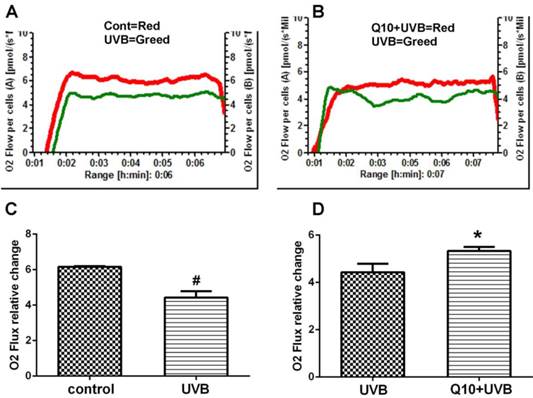
As shown in Fig. 2, the measurement of mitochondrial oxygen consumption revealed that UVB induced a 30% deduction in oxygen consumption comparing to control (P < 0.01), suggesting a suppression of mitochondrial respiratory function. Treatment of CoQ10 at 25 μg/ml improved mitochondrial respiration in UVB challenged astrocytes.
Mitochondrial respiratory rate after CoQ10 treatment in UVB exposed astrocytes. (A) Comparison between control and UVB-challenged cells. (B) Comparison between UVB and CoQ10+UVB treated cells. (C and D) Summarized data from 3 independent experiments. One-way ANOVA followed by Tukey's test. #P < 0.01 vs. control and *P < 0.05 vs. UVB. Data are presented as means ± S.D.
Production of superoxide radicals and changes of mitochondrial membrane potential
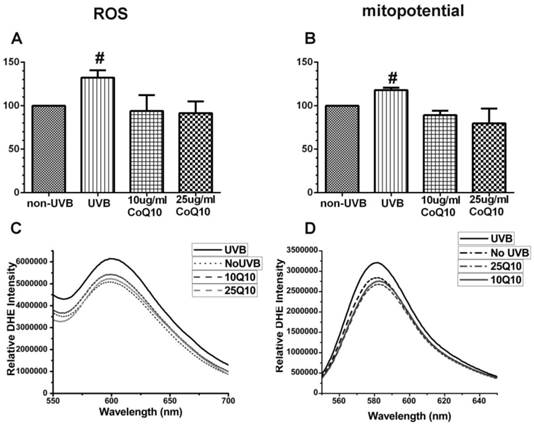
As shown in Fig. 3 left panel, UVB irradiation enhanced the production of ROS (P < 0.05) and pretreatment with 10 and 25 μg/ml CoQ10 prevented the UVB-induced ROS increase. Thus, the levels of ROS were the same in CoQ10 treated cells as in non-UVB irradiated controls. Similarly, the mitochondrial membrane potential (Fig. 3, right panel), which was measured using TMRM, increased in UVB-irradiated astrocyte cells as compared to normal control cells (P < 0.05), indicating mitochondrial membrane hyperpolarization. Treatment with CoQ10 prevented the UVB-induced mitochondrial membrane hyperpolarization. Therefore, the mitochondrial membrane potential in UVB plus CoQ10 treated cells were at the same level as in control cells (P > 0.05).
Measurements of ROS production by DHE (A, C) and mitochondrial membrane potential by TMRM (B, D). Top panels are bar graphs showing summarized ROS and mitochondrial membrane potential changes and bottom panels are representative recordings of ROS and mitochondrial membrane potential measurements, respectively. Data were collected form 3 independent experiments and presented as means ± S.D.. *P < 0.05 against control. One-way ANOVA followed by Tukey's test.
Levels of Cyto C, Caspase-9 and caspase-8
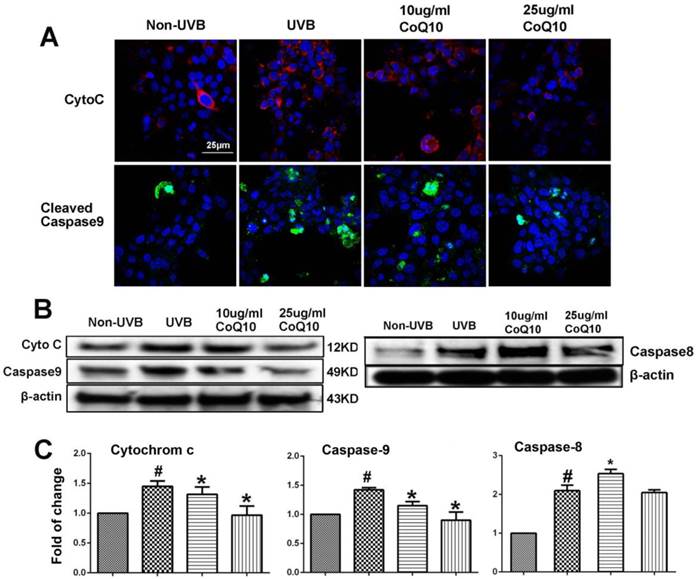
To examine whether CoQ10 inhibits mitochondria-initiated or receptor-mediated apoptotic cell death pathways, we performed Western blotting and/or immunocytochemistry using specific antibodies against cyto-c and caspase-9, and caspase-8. As shown in Fig. 4A, number of cells with cytosolic localization of cyto-c increased after UVB irradiation and decreased in cells treated with 25 µg/ml Co-Q10. Similarly, number of caspase-9 positively stained cells increased in UVB irradiated cell and decreased by Co-Q10 treatment. These results were further confirmed by protein electrophoresis. Thus, protein levels of cyto-c, cleaved caspase-9 and caspase-8 were significantly increased after UVB irradiation; whereas pretreatment with CoQ10 for 24 h at the concentrations of 25 µg/ml markedly suppressed the release of cyto-c and activation of capase-9, but not caspase-8, in the cytosolic fraction (Fig. 4B, 4C). The inhibiting effects of CoQ10 on these proapoptotic factors exhibited a dose dependent manner. When the concentration of CoQ10 increased from 10 to 25 µg/ml, the levels of the detected indices were further decreased.
Influence of UVB and effects of CoQ10 on Bax, Cyto C and caspase-9 levels. (A) Immunocytochemistry of cyto-c and cleaved caspase-9. Number of cyto-c and cleaved caspase-9 positive cells increased after UVB irradiation compared to control (Non-UVB). Pretreatment with CoQ10 at 25ug/ml significantly reduced the number of cyto-c and caspase-9 positive cells. Red color labels cyto-c, green color cleaved caspase-9, and blue color denotes DAPI stained nuclei. (B) Western blotting showing cyto-c, cleaved caspase-9 and caspase-8 in the cytoplasm collected from four experimental groups. Beta-actin used as an internal protein loading control. (C) Bar graphs summarize the protein band intensities of cyto-c, cleaved caspase-9 and caspase-8. #P < 0.01 vs. non-UVB control and *P <0.05 vs. UVB. One-way ANOVA followed by Tukey's test. Data are presented as means ± S.D.
Mitochondrial biogenesis assay. Mitochondrial biogenesis was measured using the ratio of mtDNA encoded COX-I and nDNA encoded SDH-A levels. CoQ10 increased mitochondrial biogenesis comparing to control cells. Average time course is presented in the left and a summarized bar graph in the right. #P < 0.01, Student t test. Data are presented as means ±S.D.
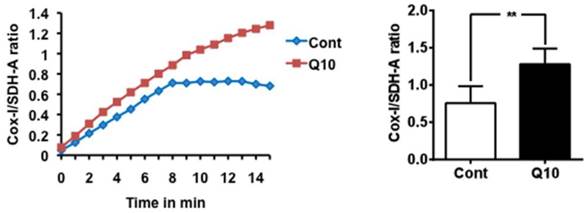
CoQ10 activates mitochondrial biogenesis
Mitochondrial biogenesis assay using the Abcam MitoBiogeneis In-Cell ELISA kit revealed an increase in the ratio of mitochondrial encoded COX-I to nuclear-DNA encoded SDH-A proteins, suggesting that CoQ10 activated mitochondrial biogenesis compared with control cells.
Discussion
CoQ10 has been shown to protect neuronal cells from UVB and ROS induced damages and to improve neurological behavior in in vivo animal models of Parkinson's disease [15,19]. It is not until recently the effects of CoQ10 were studied in astrocytes. In the present study, we have demonstrated that UVB irradiation resulted in astrocyte cell death, suppressed mitochondrial respiratory rate, enhanced ROS accumulation, increased mitochondrial membrane potential caused release of cyto-c and activation of both caspases-9 and -8. Pretreatment with CoQ10 ameliorated UVB-induced cell death, improved mitochondrial respiration, reduced ROS level, stabilized mitochondrial membrane potential, and prevented the release of cyto-c and increases of caspase-9. In the present study, we selected UVB stressed astrocytes as our experimental model. This is based on the considerations that UV irradiation has been used as an ROS stress model because the major damage mechanism of UVB is the increase in ROS production and that astrocytes in optic nerve head get direct UV irradiation. Therefore, UVB irradiated astrocytes is a clinically relevant model.
ROS are mainly produced in the mitochondria under physiological conditions. However, the physiological content of ROS is degraded by intrinsic antioxidants such as superoxide dismutases and glutathione peroxidases. Under pathological conditions, such as UVB irradiation, excessive ROS are produced, exceeds the endogenous antioxidant defense capacity [20]. It is known that hyperpolarization of mitochondrial membrane potential enhances ROS production and the latter causes damage to the mitochondria, lipid membranes, proteins and DNA. In this study, we observed an increase of mitochondrial membrane potential and elevation of ROS after UVB irradiation. Consistently, mitochondrial respiration was decreased in UVB exposed cell, suggesting UVB damages the mitochondria. CoQ10 successfully stabilized the mitochondrial membrane potential in UVB exposed cells, reduced ROS production and improved mitochondrial respiration. These suggest that CoQ10 protects the mitochondria from oxidative damage.
Cell death could be resulted from action of either intrinsic or extrinsic cell death pathways, or both. The intrinsic cell death pathway is activated by mitochondrial membrane damage, followed by release of cyto-c from the mitochondria to the cytoplasm, activation of caspase-9 and downstream caspase-3; while the extrinsic pathway is activated when a ligand binds to its receptor causing activation of caspase-8. Therefore, caspase-9 and -8 have been used to distinguish the activation of intrinsic (mitochondria-mediated) and extrinsic (receptor-mediated) cell death pathways. In the present study, protein levels of both cleaved caspases-9 and -8 increased after UVB irradiation, suggesting that UVB contributes to cell death through the activation of both the intrinsic and extrinsic cell death pathways. Co-Q10 blocks the release of cyto-c and activation of caspase-9, but failed to block activation of caspase-8, which suggest that CoQ10 inhibits the intrinsic but no the extrinsic cell death pathway.
Mitochondrial biogenesis is a highly regulated process responsible for synthesis of new mitochondria. Mitochondrial biogenesis is a promising therapeutic target to tackle mitochondrial dysfunction and bioenergetic failure during pathological conditions including ageing, neurodegeneration and stroke. Recent studies have demonstrated that CoQ10 participates in several aspects of cell signaling and affects the expression of genes involved in metabolism, cell signaling and transport [21,22]. Moreover, CoQ10 induces the AMP-activated protein kinase (AMPK)-mediated induction of PPARα, which is a known factor activating mitochondrial biogenesis, at both the mRNA and protein levels (33). In this study, we have employed the mitochondrial biogenesis kit to examine whether Co-Q10 activates mitochondrial biogenesis. The result shows that CoQ10 increased the ratio of mitochondrial DNA encoded COX-I and nuclear-DNA encoded SDH-A increased significantly in CoQ10 treated cells. This suggests that CoQ10 is capable of activating mitochondrial biogenesis. This result is consistent with the recent finding that CoQ10 increases the level of peroxisome proliferator-activated receptor (PPAR) coactivator-1α (PGC-1α), which is a well-known activator of the mitochondrial biogenesis [1].
In summary, our data have shown that the protective effect of CoQ10 is associated with its ability to decrease ROS production, to stabilize mitochondrial membrane potential, to improve mitochondrial respiration, to inhibit mitochondria-mediated cell death pathway, and to activate mitochondrial biogenesis.
Abbreviations
CoQ10, Coenzyme Q10; COX, cyclooxygenase; Cyto-c, cytochrome c; DHE, dihydroethidine; DMEM, Dulbecco's modified Eagle's medium; FBS, Fetal bovine serum; MTT, 3-(4, 5,-dimethythiazol-2-yl)-2, 5-diphenyl tetrazolium bromide; PBS, phosphate buffered saline; PPAR, peroxisome-proliferator-activated receptor; PGC-1α, PPAR-γ coactivator-1; RFI, relative fluorescence intensity; SDH-A, succinate dehydrogenase complex subunit A; TMRM, tetramethylrhodaminemethyl ester; UV, Ultraviolet; UVB, Ultraviolet B.
Acknowledgements
LJ was supported by National Science Foundation of China (81260184) and Ningxia Medical University Research Fund (XT2011009, KF20100-26); JZZ by the National Science Foundation of China (81360184); and PAL by the National Institute of Health (7R01DK075476-06). The authors greatly appreciate Dr. Carla Oldham at the North Carolina Central University for proofread the manuscript.
Authors' contributions
Conceived and designed the experiments: LJ, JZZ, PAL. Performed the experiments: LJ, MH, YC, QPH, SLM. Contributed reagents/materials/analysis tools: PAL, MH. Wrote the paper: LJ, MH, JZZ, PAL.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Noh YH, Kim KY, Shim MS, Choi SH, Choi S, Ellisman MH, Weinreb RN, Perkins GA, Ju WK. Inhibition of oxidative stress by coenzyme Q10 increases mitochondrial mass and improves bioenergetic function in optic nerve head astrocytes. Cell Death and Disease. 2013;4(10):e820
2. Molofsky AV, Krenick R, Ullian E. et al. Astrocytes and disease: a neurodevelopmental perspective. Genes Dev. 2012;26:891-907
3. Gosejacob D, Dublin P, Bedner P, Hüttmann K, Zhang J, Tress O, Willecke K, Pfrieger F, Steinhäuser C, Theis M. Role of astroglial connexin30 in hippocampal gap junction coupling. Glia. 2011;59(3):511-9
4. Barnett SC, Linington C. Myelination: do astrocytes play a role? Neuroscientist. 2013;19(5):442-50
5. Tsai HH, Li H, Fuentealba LC, Molofsky AV, Taveira-Marques R, Zhuang H, Tenney A, Murnen AT, Fancy SP, Merkle F, Kessaris N, Alvarez-Buylla A, Richardson WD, Rowitch DH. Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science. 2012;337(6092):358-62
6. Cho KS, Kwon KJ, Jeon SJ, Joo SH, Kim KC, Cheong JH, Bahn GH, Kim HY, Han SH, Shin CY, Yang SI. Transcriptional Upregulation of Plasminogen Activator Inhibitor-1 in Rat Primary Astrocytes by a Proteasomal Inhibitor MG132. Biomol Ther (Seoul). 2013;21(2):107-13
7. Chaturvedi R, Reddig K, Li HS. Long-distance mechanism of neurotransmitter recycling mediated by glial network facilitates visual function in Drosophila. Proc Natl Acad Sci. 2014;111(7):2812-7
8. Muranyi M, Ding C, He Q, Lin Y, Li PA. Streptozotocin-induced diabetes ca8uses astrocyte death after ischemia and reperfusion injury. Diabetes. 2006;55(2):349-55
9. Jing L, He Q, Zhang JZ, Li PA. Temporal profile of astrocytes and changes of oligodendrocyte-based myelin following middle cerebral artery occlusion in diabetic and non-diabetic rats. Int J Biol Sci. 2013;9(2):190-9
10. Vincent AM, Olzmann JA, Brownlee M, Sivitz WI, Russell JW. Uncoupling proteins prevent glucose-induced neuronal oxidative stress and programmed cell death. Diabetes. 2004;53(3):726-34
11. Alfonso-Loeches S, Ureña-Peralta JR, Morillo-Bargues MJ, Oliver-De La Cruz J, Guerri C. Role of mitochondria ROS generation in ethanol-induced NLRP3 inflammasome activation and cell death inastroglial cells. Front Cell Neurosci. 2014;8:216
12. Hsieh HL, Chi PL, Lin CC, Yang CC, Yang CM. Up-regulation of ROS-Dependent Matrix Metalloproteinase-9 from High-Glucose-Challenged Astrocytes Contributes to the Neuronal Apoptosis. Mol Neurobiol. 2014 [Epub ahead of print]
13. Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283(2-3):65-87
14. Fukui M, Zhu BT. Mitochondrial superoxide dismutase SOD2, but not cytosolic SOD1, plays a critical role in protection against glutamate-induced oxidative stress and cell death in HT22 neuronal cells. Free Radic Biol Med. 2010;48:821-830
15. Sikorska M, Lanthier P, Miller H, Beyers M, Sodja C, Zurakowski B, Gangaraju S, Pandey S, Sandhu JK. Nanomicellar formulation of coenzyme Q10 (Ubisol-Q10) effectively blocks ongoing neurodegeneration in the mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model: potential use as an adjuvant treatment in Parkinson's disease. Neurobiol of Aging. 2014;35(10):2329-2346
16. Nucci C, Tartaglione R, Cerulli A, Mancino R, Spanò A, Cavaliere F, Rombolà L, Bagetta G, Corasaniti MT, Morrone LA. Retinal damage caused by high intraocular pressure-induced transient ischemia is prevented by coenzyme Q10 in rat. International Review of Neurobiology. 2007;82:397-406
17. Jing L, Kumari S, Mendelev N, Li PA. Coenzyme Q10 ameliorates ultraviolet B irradiation induced cell death through inhibition of mitochondrial intrinsic Cell Death Pathway. International Journal of Medical Science. 2011;12(11):8302-8315
18. Li HN, Chen GS, Ma WR, Li PA. Water-Soluble Coenzyme Q10 Inhibits Nuclear Translocation of Apoptosis Inducing Factor and Cell Death Caused by Mitochondrial Complex I Inhibition. Int. J. Mol. Sci. 2014;15:13388-13400
19. Yang L, Calingasan NY, Wille EJ, Cormier K, Smith K, Ferrante RJ, Beal MF. Combination therapy with coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson's and Huntington's diseases. J Neurochem. 2009;109(5):1427-39
20. Ryan JM, Harper ME. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radical Biology and Medicine. 2011;51(6):1106-1115
21. Groneberg DA, Kindermann B, Althammer M, Klapper M, Vormann J, Littarru GP, Döring F. Coenzyme Q10 affects expression of genes involved in cell signaling, metabolism and transport in human CaCo-2 cells. Int J Biochem Cell Biol. 2005;37:1208-1218
22. 22.Littarru GP, Tiano L. Bioenergetic and antioxidant properties of coenzyme Q10: recent developments. Mol Biotechnol. 2007;37:31-37
Author contact
![]() Corresponding authors: Dr. Jian-Zhong Zhang, email: zhangjzedu.cn; or Dr. P. Andy Li, email: pliedu
Corresponding authors: Dr. Jian-Zhong Zhang, email: zhangjzedu.cn; or Dr. P. Andy Li, email: pliedu