Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(7):803-812. doi:10.7150/ijbs.11100 This issue Cite
Research Paper
PERK-mediated Autophagy in Osteosarcoma Cells Resists ER Stress-induced Cell Apoptosis
Guang-rong Ji ![]() , Nai-chun Yu, Xiang Xue, Zong-guang Li
, Nai-chun Yu, Xiang Xue, Zong-guang Li
Department of Orthopaedics, The 2nd Affiliated Hospital of Harbin Medical University, Harbin, 150001, China
Received 2014-11-18; Accepted 2015-2-27; Published 2015-5-23
Abstract
Osteosarcoma is a bone cancer that develops commonly in children and adolescents. However, osteosarcoma treatments often fail by the development of chemoresistance to apoptosis, and the molecular mechanisms remain unclear. In this study, we propose that autophagy is responsible for osteosarcomatous resistance to apoptosis. We implicate PERK-mediated autophagy as a significant contributor to apoptosis resistance due to ER stress in osteosarcoma cells. By immunostainings and western blots, we identified that PERK activated osteosarcomatous autophagy via inhibiting mTORC1 pathway, thereby preventing cell apoptosis. While using RNAi, we knocked down PERK and found that autophagy was suppressed, result in osteosarcomatous apoptosis. Our results identify a novel role of PERK-mediated autophagy as a significant mechanism for osteosarcoma cell survival. These results will help to understand the mechanism of chemoresistance in osteosarcoma cells, and indicate a novel target for improving osteosarcoma therapy.
Keywords: PERK, osteosarcoma, autophagy, ER stress, mTORC1
Introduction
Osteosarcoma (OS) is a type of bone tumor in which the proliferating spindle cells produce osteoid or immature bone. OS accounts for 2.4 % of all malignancies in pediatric patients and about 20 % of primary bone cancers [1, 2]. OS occurs mainly in the metaphysis of long bones around the knee region of the distal femur or proximal tibia. It is highly aggressive and metastasizes mainly to the lung [3]. Recently, advances in clinical treatment have helped much to improve limb salvage and reduce metastases. And multiagent dose-intensive chemotherapy methods have increased the disease-free survival rates in patients with localized disease [4]. However, frequent acquisition of drug-resistant phenotypes is often found in OS chemotherapy. It becomes a significant obstacle to develop better OS clinical treatment. With drug treatment, osteosarcoma cells activate several intracellular protective mechanisms to escape from cell death [5, 6]. Earlier reports have been indicated that some strategies are used by osteosarcoma cells to obtain apoptosis resistance, such as PI3K/AKT, MDR-I and survivin pathways [7-10]. Although much progress has been made, it is still urgent to clarify molecular mechanisms underlying OS chemoresistance for clinical therapy.
In mammalian cells, many mechanisms that modulate cell homeostasis developed to maintain the balance of self construction and destruction. And one of these mechanisms is autophagy [11]. Autophagy is a fundamental lysosomal process that participates in stress tolerance. By autophagy, impaired/disrupted proteins and organelles are recruited to autophagosomes and subsequently degraded by enzymes. It is because of autophagy that intracellular components may be recycled to maintain homeostasis and prevent the accumulation of damaged cell fragements [12]. Multiple stressful conditions may induce autophagy, like ER stress. ER stress has been reconsidered as an imbalance between protein synthesis and processing [13]. When ER stress is overwhelmed, cells will initiate autophagy, and further lead to large-scale degradation and apoptosis [14]. Thus, autophagy may serve as a protective mechanism against cell stress [15-17]. Actually, autophagy occurs at basal levels in normal tissues. However, it is ectopically activated in tumor cells, conferring to chemoresistance [18-20]. Although autophagy has been identified as a protective mechanism against stress in many tumor cells, the definite mechanism and significance of autophagy in tumor chemoresistance, especially in OS, remains largely unknown.
To study the role of autophagy on chemotherapy resistance, we proposed to establish a molecular link between ER stress, autophagy and apoptosis in osteosarcoma cells. PERK is a ubiquitously-expressed ER protein kinase, which phosphorylates the alpha subunit of eIF2α and plays a significant role in tumor development [21, 22]. When the protein folding capacity of ER is compromised during UPR, the canonical function of PERK is to release the protein load in the ER, thereby attenuating translation initiation [23]. So PERK-knockout cells are unable to modulate ER client protein load and experience extremely high levels of ER stress [24].
In the present study, we firstly demonstrated that PERK is highly expressed in MG63 human osteosarcoma cells and clinical osteosarcoma samples. Loss of PERK may enhance the UPR and ER stress under both basal and ER stress conditions. Moreover, PERK knockdown was found to induce more dramatic cell apoptosis than those in the controls, which may be due to autophagy inhibition. And the inhibited autophagy in PERK-KD cells may be caused by ectopic activation of mTOR pathway. We also found that re-activation of autophagy in PERK-KD cells may rescue cell apoptosis induced by ER stress. Our results have established a molecular network of ER stress, autophagy and apoptosis in osteosarcoma cells, and characterized the role of autophagy in the resistance to osteosarcomatous apoptosis. These findings will allow us to develop new therapeutic strategies for clinical treatments of osteosarcoma.
Materials and Methods
Chemicals and materials
Thapsigargin was purchased from Sigma-Aldrich (St. Louis, MO, USA). Rapamycin was from Invitrogen (Carlsbad, CA, USA). DMEM and FBS were purchased from GIBCO Invitrogen (Carlsbad, CA, USA). The Hoechst kit and Lyso-Tracker Red probe for acidic lysosome staining were from Beyotime (Haimen, Jiangsu, China). Anti-PERK, Anti-BiP, anti-p-eIF2α, anti-eIF2α, anti-cleaved-caspase 3, anti-LC3, anti-p-AKT, anti-p-p70S6K1, anti-p70S6K1, anti-p-4EBP1 and anti-4EBP1 were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-ATF-6, anti-p62 and anti-GAPDH were from Abcam (Cambridge, UK). The CHOP antibody was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All other reagents were obtained from Sigma-Aldrich with the highest purity available.
Assay of cell cultures and pharmacological manipulations
Human osteosarcoma cell line (MG63) was purchased from the Shanghai Institute of Cell Biology (introduced from the American Type Culture Collection). MG63 cells were incubated in DMEM containing 10 % FBS plus antibiotics at 37 °C by 1.0×106 cells/mL. For PERK knockdown, MG63 cells were transfected with siRNA heteroduplexes specific for human PERK (5'-CTCACAGGCAAAGGAAGGAG-3') [25]. After the transfection for 48 h, cells were collected for following experiments. For the primary osteoblast cell culture, the operation processes were carried out according to standard procedures by Divieti's report [26].
To induce ER stress in MG63 cells, thapsigargin was applied to the cells at a final concentration 1 μM for 6 h. Cells with an equivalent amount of DMSO were used as internal controls. Finally, cells were harvested for mRNA or protein examinations. Additionally, rapamycin treatment at 100 nM for 6 h served to induce autophagy.
Clinical samples
Ten clinical samples from osteosarcoma patients were collected for PERK expression examinations. Patients were admitted in Department of Orthopedics of the 2nd Affiliated Hospital of Harbin Medical University. Participants provided written informed consent approved by the Hospital Human Research Ethics Committee of the 2nd Affiliated Hospital of Harbin Medical University.
Cell Staining
For Hoechst staining, cells were plated in 6-well plates at 1.0×105 cells/mL. After pharmacological manipulation, cells were stained using the Hoechst kit from Beyotime. The cell counting operations were performed by the National Institutes of Health software ImageJ, which is available from http://rsbweb.nih.gov.
For immunostaining, MG63 cells were grown on glass coverslips. For GFP-LC3 puncta staining, MG63 cells were transfected with a GFP-tagged LC3 expression plasmid using Lipofectamine 2000. After transfection of 48 h, cells were fixed (plus sucrose) in PBS and permeabilized with Triton-X 100 at room temperature. After incubation with proper primary and fluorescent secondary antibodies, cells were mounted onto glass slides with an antifade reagent containing DAPI and visualized by fluorescence microscopy.
To detect the autophagy status of MG63 cells, we applied Lyso-Tracker Red staining to examine acidic lysosomes. For the assays of Lyso-Tracker Red staining, MG63 cells were plated at 1.0×105 cells/mL in 6-well plates. After thapsigargin treatment at a final concentration of 1 μM for 6 h, living cells were directly stained with Lyso-Tracker Red probes from Beyotime following the instructions.
Real-time PCR
Total RNA were extracted by traditional Trizol reagent. Subsequent reverse transcription was operated by reverse transcriptase from MBI/Fermentas. Quantitative real-time PCR was performed in Bio-Rad iQ5 machine, and GAPDH worked as the internal control. Primer sequences are shown in Table 1.
Primer sequences for SYBR Green probes of target genes
| Gene | Primer sequence | |
|---|---|---|
| Bip | forward | 5'-CGCTTCGAATCGGCGGTACCCAG-3' |
| reverse | 5'-TTCTTGTCCTCCTCCTAAGCTTCG-3' | |
| Atf6 | forward | 5'-AACAAGACCACAAGACCA-3' |
| reverse | 5'-AGGAGGAACTGACGAACT-3' | |
| Gapdh | forward | 5'-ACACCCACTCC TCCACCTTT-3' |
| reverse | 5'-TAGCCAAATTCGTTGTCATA-3' | |
Cell sample preparation and Western blot
To extract proteins, the cultured cells were harvested by centrifugation at l0, 000 rpm for 10 min. Next, cells were sonicated with SDS buffer plus protease inhibitors. Protein concentrations were examined and corrected by BCA Protein Assay kit (Thermo Fisher Scientific Inc., Rockford, IL, USA). Equal amounts of the proteins from each extract were separated on a SDS-PAGE. The following Western blots were carried out according to standard procedures. Final proteins were detected and quantified using ImageJ.
Statistical analysis
Quantitative data are shown as the mean ± SEM using ANOVA with post-hoc tests for comparisons. The p-values of 0.05 (*), 0.01 (**) and 0.001 (***) were considered as the levels of significance for the statistical tests.
Results
PERK is highly expressed in MG63 human osteosarcoma cells
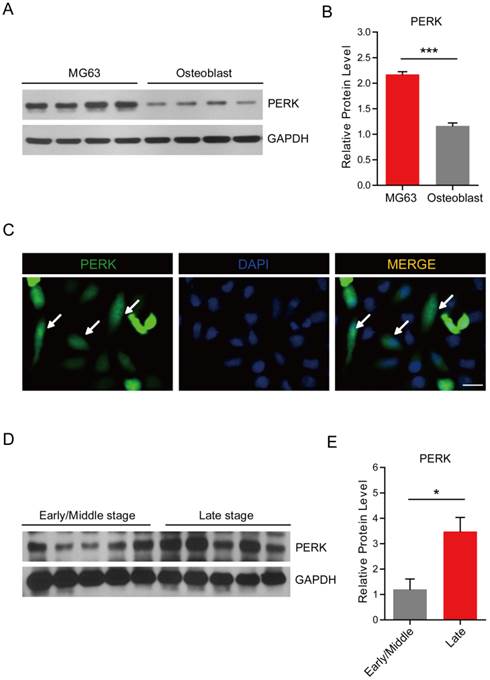
Earlier results have showed that the expression of PERK is increased in some types of tumors, and may mediate cell growth and survival under ER stress conditions [27, 28]. To determine whether osteosarcoma-derived PERK retains high expression, we assessed PERK expression in MG63 human osteosarcoma cells compared to primary osteoblasts. These biochemical results showed that the PERK protein level was higher in MG63 cells than those in osteoblasts, by approximately 2.2-folds (Fig. 1A and B). To further validate the expression pattern of PERK in osteosarcoma cells, we carried out PERK immunostaining to directly visualize PERK localization in MG63 cells. Green fluorescence indicated that PERK was widely expressed in the both cytosol and nuclei of MG63 cells, which was consistent with previous reports of other types of tumor cells (Fig. 1C). Furthermore, to detect the expression of PERK in clinical osteosarcoma tissues, we assessed the PERK protein level in different stages of osteosarcoma patients. Results show that PERK expression was increased by approximately 3.5-fold in the late stages of osteosarcoma compared to that in the early/middle stages (Fig. 1D and E). Taken together, these results suggest that PERK is highly expressed in osteosarcoma cells, which may play an important role in osteosarcoma development and survival.
PERK is highly expressed in MG63 human osteosarcoma cells. (A-B) Western blots and histograms showing the high expression of PERK in MG63 osteosarcoma cells compared to primary osteoblasts. Results are the average of four independent experiments. Data represent mean ± SEM. ***p<0.001. (C) Images showing the expression pattern of PERK in cultured MG63 cells. Green fluorescence indicates PERK, and blue indicates DAPI. Bar 25 μm. (D-E) Western blots and histograms showing that PERK expression is higher in late stages of osteosarcoma than in early/middle stage samples. Results are the average of five pairs of clinical samples. Data represent mean ± SEM. *p<0.05.
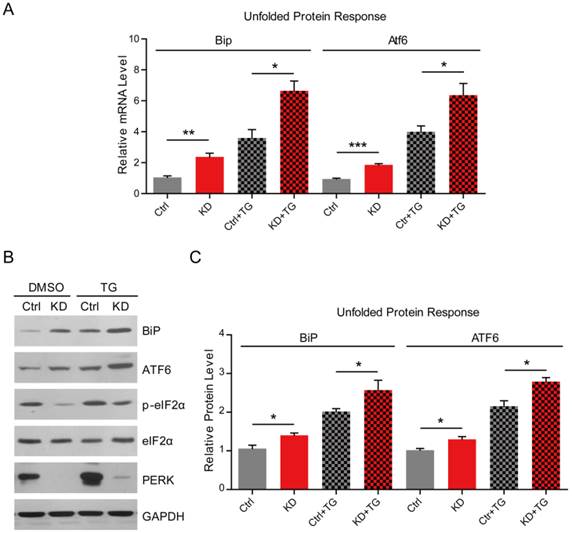
PERK knockdown enhances ER stress in MG63 human osteosarcoma cells
To examine whether osteosarcomatous ER stress is upregulated by PERK loss, we investigated the gene expression of UPR in both control and PERK-knockdown osteosarcoma cells. Real-time PCR results show that mRNA levels of Bip and Atf6, which are canonical markers of the UPR, increased by 2.3 and 1.9-folds in PERK-KD cells (Fig. 2A). Moreover, when thapsigargin (TG) was applied to induce ER stress, the mRNA levels of Bip and Atf6 in PERK-KD cells increased more dramatically (Bip-6.6 and Atf6-6.4 folds) than those in wild type cells (Bip 3.6-fold and Atf6 4.0-fold) (Fig. 2A). To further confirm the increased ER stress by PERK-KD in MG63 cells, we examined the protein levels of UPR markers BiP, ATF6 and p-eIF2α. The protein levels of these UPR markers were increased by 1.4-(basal BiP), 1.3-(basal ATF6), 2.6-(TG BiP) and 2.8-folds (TG ATF6) in PERK-KD cells, compared to 2.0-(TG BiP) and 2.1-folds (TG ATF6) in wild type cells (Fig. 2B and C). These results clearly indicated that knockdown of PERK may increase the ER stress sensitivity in osteosarcoma cells, and leads to heavy loads in the ER. Notably, as the downstream indicators of PERK, we noticed that p-eIF2α protein level was reduced under both basal and TG conditions, confirming PERK inactivation (Fig. 2B and C). Based on the above results, we identify that knockdown of PERK may enhance ER stress in osteosarcoma cells, which may lead to cell apoptosis.
PERK knockdown enhances ER stress in MG63 human osteosarcoma cells. (A) Real-time PCR results showing the increasing of UPR genes Bip and Atf6 in MG63 cells by PERK knockdown under both basal and thapsigargin (1 μM for 6 h) conditions. Results are the average of three independent experiments. Data represent mean ± SEM. *p<0.05, **p<0.01 and ***p<0.001. (B-C) Western blots and histograms showing the increasing of BiP and ATF6 protein levels by PERK knockdown under both basal and thapsigargin (1 μM for 6 h) conditions. Note that the protein level of p-eIF2α/eIF2α was reduced by PERK knockdown. Results are the average of three independent experiments. Data represent mean ± SEM. *p<0.05.
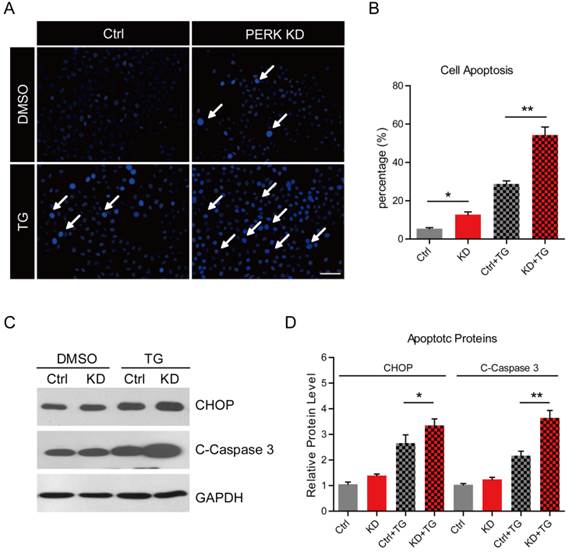
PERK knockdown promotes ER stress-mediated apoptosis in MG63 human osteosarcoma cells
The effects of ER stress on cell apoptosis differs in different types of tumor cell lines. To examine the effect of ER stress on cell apoptosis in osteosarcoma cells, we carried out Hoechst staining to visualize the cell status. The results showed that, under basal conditions, the percentages of apoptotic cells were slightly increased in PERK-KD cells compared to those in wild type cells. However, after thapsigargin treatment, we noted the percentages of apoptotic cells reached 28.8 % in wild type MG63 cells and was extremely high (54.3 %) in PERK-KD cells (Fig. 3A and B). This finding is consistent with the alternations of UPR in PERK-KD cells, suggesting that loss of PERK may reduce cell resistance to stress. To confirm these findings, we carried out western blots to analyze the apoptotic protein levels in PERK-KD MG63 cells. We found that the protein levels of CHOP and cleaved-caspase 3, well-known apoptotic proteins, were increased more in PERK-KD cells than that in wild type cells under both basal and thapsigargin conditions (Fig. 3C and D). Taken together, our findings strongly indicated that loss of PERK may remarkably reduce cell resistance in osteosarcoma cells, inducing dramatic cell death.
PERK knockdown promotes ER stress-mediated apoptosis in MG63 human osteosarcoma cells. (A) Hoechst staining (blue) showing increased cell apoptosis with thapsigargin (1 μM for 6 h) treatment in PERK knockdown MG63 cells compared to wild type MG63 cells. White arrows indicate apoptotic cells. Bar 50 μm. (B) Histograms showing the quantification of cell apoptosis (%) in wild type and PERK knockdown MG63 cells after thapsigargin treatment. Results are the average of four independent experiments. Data represent mean ± SEM. *p<0.05, **p<0.01. (C-D) Western blots and histograms showing increased protein levels of CHOP and cleaved caspase 3 (c-caspase 3) with thapsigargin treatment (1 μM for 6 h) in PERK knockdown cells compared to wild type MG63 cells. Results are the average of three independent experiments. Data represent mean ± SEM. *p<0.05, **p<0.01.
PERK knockdown reduces autophagy by activating mTORC1 pathway in MG63 human osteosarcoma cells
To study how osteosarcoma cells resist ER stress and why the loss of PERK reduces cell resistance to stress, we focused on an important cellular protective mechanism against apoptosis, autophagy. To examine whether autophagy was increased in osteosarcoma cells induced by ER stress, we applied a living lysosome staining to these cells. The Lyso-Tracker Red probe provides highly selective staining of acidic lysosomes, which are recognized as markers of autophagy [29]. By this assay, we found that the intensity of red fluorescence was increased by 8.5-fold in wild type MG63 cells after thapsigargin treatment, and only 4.3-fold in PERK-KD cells (Fig. 4A and B). To confirm these results, we carried out GFP-LC3 puncta assays to detect autophagy [30]. The images show that with GFP-LC3 transfection, many punctas appeared in wild type MG63 cells after thapsigargin treatment. However, the puncta numbers were reduced in PERK-KD cells after thapsigargin treatment (Fig. 4C). Moreover, biochemical results show that thapsigargin induced the conversion of LC3-I to LC3-II and reduced p62, both markers of autophagy, in wild type MG63 cells, but not in PERK-KD cells (Fig. 4D). Thus, our data indicates that loss of PERK disrupts the induction of autophagy by ER stress.
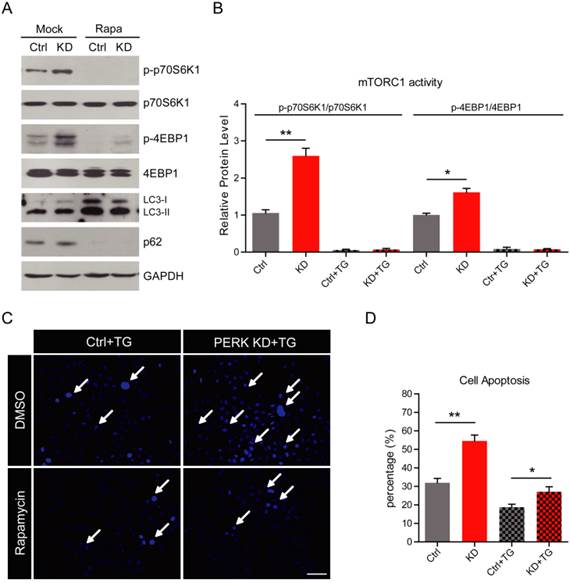
PERK knockdown reduces autophagy by activating the mTORC1 pathway in MG63 human osteosarcoma cells. (A) Acidic lysosome staining by Lyso-Tracker Red probes showing the attenuated induction of autophagy by thapsigargin (1 μM for 6 h) treatment in PERK knockdown MG63 cells. Bar 25 μm. (B) Histograms showing the quantification of the relative fluorescence intensity in wild type and PERK knockdown MG63 cells after thapsigargin treatment. Results are the average of three independent experiments. Data represent mean ± SEM. **p<0.01. (C) Images showing the induction of GFP-LC3 puncta reduced by PERK knockdown after thapsigargin (1 μM for 6 h) treatment. White arrows indicate GFP-LC3 puncta. Bar 5 μm. (D) Western blots showing the induction of autophagy (indicated by the LC3-I/II ratio and p62) and inhibition of the mTORC1 pathway (indicated by p-70S6K1 and p-4EBP1) after thapsigargin (1 μM for 6 h) treatment. PERK knockdown constitutively activates themTORC1 pathway and inhibits autophagy after thapsigargin treatment.
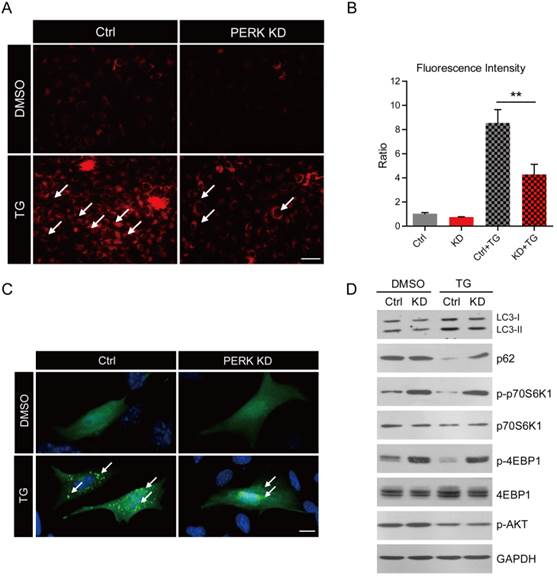
To investigate the molecular mechanism of PERK on autophagy, we focused on mTOR signaling. mTOR is a master regulator that integrates signals from growth factors and nutrients to cell metabolism [31]. The activation of mTORC1 pathway inhibits autophagy [32]. While inhibition of mTORC1 by rapamycin promotes autophagy in hepatoma and breast cancer cells [33, 34]. To study whether PERK reduces autophagy through mTORC1 pathway, we examined the activity of mTORC1 in osteosarcoma cells. We found that p-p70S6K1 and p-4EBP1, which are markers of mTORC1 pathway, were dramatically increased by PERK knockdown under both basal and thapsigargin conditions (Fig. 4D). We also found that mTORC2 activity, indicated by p-AKT, may not be altered by PERK-KD (Fig. 4D). These results suggest that ectopic activation of the mTORC1 pathway may be responsible for autophagy inhibition in PERK-KD osteosarcoma cells.
Inhibition of the mTORC1 pathway promotes cell survival in PERK-knockdown MG63 cells
We have shown that PERK loss may block ER stress-mediated autophagy and result in cell apoptosis in MG63 cells. Thus, it highly possible that re-activation of autophagy in PERK-KD cells may restore cell survival in osteosarcoma cells. To test this hypothesis, we applied autophagy activators to thapsigargin-treated PERK-KD cells and examined cell viability. Rapamycin is a well-known mTORC1 inhibitor, and widely used as an autophagy inducer. After rapamycin treatment, we found that mTORC1 activity, indicated by the expressions of p-p70S6K1 and p-4EBP1, was dramatically reduced in both wild type and PERK-KD MG63 cells under thapsigargin conditions (Fig. 5A and B). Moreover, the increased expression and conversion of LC3 and decreased p62 expression indicated increased autophagy in both wild type and PERK-KD MG63 cells (Fig. 5A). Further results of Hoechst staining showed that the percentages of apoptotic cell were dramatically reduced to 27.1 % in rapamycin-treated PERK-KD cells, compared to 54.5 % in non-rapamycin PERK-KD cells (Fig. 5B and C). Therefore, we demonstrate that re-activation of autophagy may protect PERK-KD osteosarcoma cells from apoptosis, and confirm the role of autophagy in osteosarcoma resistance to ER stress.
Inhibition of the mTORC1 pathway promotes cell survival in PERK-knockdown MG63 cells. (A-B) Western blots and histograms showing that inhibition of the mTORC1 pathway by rapamycin (100 nM for 6 h) dramatically blocks mTORC1 activity (indicated by p-70S6K1 and p-4EBP1) and induces autophagy (indicated by LC3-I/II ratio and p62) in both wild type and PERK knockdown MG63 cells after thapsigargin treatment. Results are the average of three independent experiments. Data represent mean ± SEM. *p<0.05, **p<0.01. (C) Hoechst staining (blue) showing that inhibition of mTORC1 pathway by rapamycin dramatically reduced apoptosis in PERK knockdown MG63 cells after thapsigargin treatment. White arrows indicate apoptotic cells. Bar 50 μm. (D) Histograms showing the quantification of cell apoptosis (%) in wild type and PERK knockdown MG63 cells after rapamycin/thapsigargin treatment. Results are the average of four independent experiments. Data represent mean ± SEM. *p<0.05, **p<0.01.
Discussion
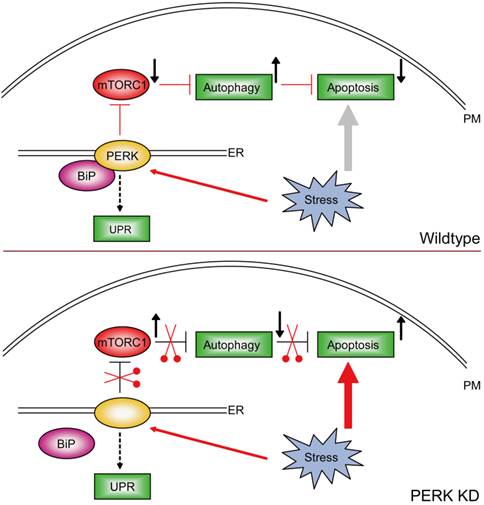
OS patients often develop resistance to chemotherapeutic drugs, and some mechanisms have been revealed for this chemoresistance. In this study, our results showed that PERK-activated autophagy is a significant contributor to the resistance of ER stress and/or apoptosis in osteosarcoma cells. PERK may inhibit mTORC1 activity and thus induce autophagy under stress conditions. Knockdown of PERK disrupted the axis of the PERK/mTORC1/autophagy and increased the osteosarcomatous sensitivity to ER stress-mediated apoptosis (Fig. 6).
Model. Schematic representation highlighting the molecular link between ER stress, autophagy and apoptosis in MG63 human osteosarcoma cells. To release cellular stress, osteosarcoma-derived PERK inactivates the mTORC1 pathway and develop autophagy to prevent cell apoptosis. With the loss of PERK, the mTORC1 pathway is constitutively activated. Therefore, osteosarcoma cells fail to induce autophagy and thus undergo dramatic cell apoptosis.
PERK is a transmembrane serine/threonine protein kinase that phosphorylates two known substrates: the eIF2α [21] and the Nrf2 transcription factor [35]. Importantly, PERK is required for maintaining cellular redox homeostasis and preventing ROS-induced genotoxic stress [23]. Recent investigations reveal PERK functions during tumorigenesis. For example, PERK promotes tumor proliferation and growth by limiting oxidative DNA damage [27]. PERK is also critical for tumor adaptation to hypoxic stress by modulating angiogenesis [36, 37]. Here, for the first time, we show that PERK is highly expressed in osteosarcoma cells, and functions as a stress responder to promote cellular protective mechanisms, such as autophagy. Therefore, our findings at least revealed that besides the functions in tumor growth and the ROS response, PERK may be an important osteosarcomatous protective gene.
Cells have an intrinsic mechanism of apoptosis under stress conditions, but tumor cells often have multiple signaling pathways and effector mechanisms to deal with temporal and developmental cell stress. Resistance to apoptosis may augment the escape of tumor cells from surveillance by the immune system. When cellular stress is overwhelmed, tumor cells may activate many protective pathways to release the stress and escape from apoptosis. During these tumorous responses to stress, autophagy plays an important role. This might partly answer why osteosarcoma cells are “stronger” than normal cells under stress conditions and drug treatment. Nowadays, chemoresistance constitutes an important clinical problem especially in OS therapy [38]. In present study, we proposed a novel mechanism of PERK-mediated autophagy in osteosarcomatous resistance to apoptosis. Our findings will help to better understand the relationship between ER stress, autophagy and apoptosis, and allow us to develop new therapeutic strategies for clinical osteosarcoma treatment.
On the other hand, the balance between new protein synthesis and unfolded protein degradation might be precisely regulated in cell maintenance. Interestingly, we noted that thapsigargin may induce ER stress in osteosarcoma cells. It also exhibited an inhibitory effect on mTORC1 pathway, which may reduce new protein synthesis. Since mTORC1 works as a master regulator of protein synthesis, controlling ~15-20% new protein synthesis in cells [31], the inhibitory effect of thapsigargin on protein synthesis may be partly mediated by mTORC1 pathway. Additionally, thapsigargin may also induce protective autophagy by inhibition of the mTORC1 pathway, which may help to release overwhelming ER stress. The co-interactions of new protein synthesis and autophagy promote osteosarcoma survival under ER stress. With the loss of PERK, the protective mechanisms of inhibited protein synthesis and activated autophagy are blocked due to constitutively activated mTORC1 pathway. Hence, we propose that PERK is a key stress sensor in osteosarcoma cells. PERK inhibitors, such as GSK2656157, could be targeted in OS clinical therapy in the future [39].
Conclusion
In summary, our results demonstrate that PERK regulates osteosarcomatous autophagy through mTORC1 pathway under ER stress, thereby preventing cell apoptosis. Loss of PERK in osteosarcoma cells suppresses protective autophagy, whereas it increases sensitivity to stress and may result in osteosarcomatous apoptosis. In the context of therapeutic intervention, inhibition of PERK may result in a highly effective novel therapeutic approach when combined with agents that induce cell apoptosis during osteosarcoma clinical therapy.
Abbreviations
ATF-6: Activating Transcription Factor 6; AKT: Protein Kinase B; BiP: Binding Protein; BCA: Bicinchoninic Acid; CHOP: C/EBP Homologous Protein; DAPI: 4'-6-diamidino-2-phenylindole; DMEM: Dulbecco's Modified Eagle's Medium; DMSO: Dimethyl Sulfoxide; 4EBP1: eIF4E-binding protein 1; eIF2α: protein Eukaryotic Initiation Factor 2α; ER: Endoplasmic Reticulum; GFP: Green Fluorescent Protein; GAPDH: Glyceraldehyde-3-phosphate Dehydrogenase; GSK: Glycogen Synthase Kinase; FBS: Fetal Bovine Serum; LC3: Light Chain 3; MDR-I: Multidrug Resistance-I; mTOR: Mammalian Target of Rapamycin; Nrf2: Nuclear Factor (erythroid-derived 2)-like 2; OS: Osteosarcoma; p-AKT: phospho-Protein Kinase B; p70S6K1: P70 ribosomal protein S6 Kinase 1; p-eIF2α: phospho-protein Eukaryotic Initiation Factor 2α; PERK: Protein kinase R (PKR)-like Endoplasmic Reticulum Kinase; PERK-KD: PERK Knockdown; PI3K: Phosphoinositide 3-kinase; ROS: Reactive Oxygen Species; SDS: Sodium Dodecyl Sulfate; UPR: Unfolded Protein Response.
Acknowledgements
This work was supported by grants from Harbin special funds for technological innovation research project (No. 2012RFQXS056).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mutsaers AJ, Walkley CR. Cells of origin in osteosarcoma: mesenchymal stem cells or osteoblast committed cells? Bone. 2014;62:56-63
2. Savage SA, Mirabello L, Wang Z. et al. Genome-wide association study identifies two susceptibility loci for osteosarcoma. Nature Genet. 2013;45:799-803
3. Broadhead ML, Clark JC, Myers DE. et al. The molecular pathogenesis of osteosarcoma: a review. Sarcoma. 2011;2011:959248
4. Meyers PA, Schwartz CL, Krailo MD. et al. Osteosarcoma: the addition of muramyl tripeptide to chemotherapy improves overall survival--a report from the Children's Oncology Group. J Clin Oncol. 2008;26:633-8
5. Brambilla D, Zamboni S, Federici C. et al. P-glycoprotein binds to ezrin at amino acid residues 149-242 in the FERM domain and plays a key role in the multidrug resistance of human osteosarcoma. Int J Cancer. 2012;130:2824-34
6. Wunder JS, Bull SB, Aneliunas V. et al. MDR1 gene expression and outcome in osteosarcoma: a prospective, multicenter study. J Clin Oncol. 2000;18:2685-94
7. Zhang B, Shi ZL, Liu B. et al. Enhanced anticancer effect of gemcitabine by genistein in osteosarcoma: the role of Akt and nuclear factor-kappaB. Anti-cancer Drugs. 2010;21:288-96
8. Tsai HC, Huang CY, Su HL. et al. CTGF increases drug resistance to paclitaxel by upregulating survivin expression in human osteosarcoma cells. Biochimica Biophys Acta. 2014;1843:846-54
9. Wang G, Rong J, Zhou Z. et al. Novel gene P28GANK confers multidrug resistance by modulating the expression of MDR-1, Bcl-2 and Bax in osteosarcoma cells. Mol. Biol. (Mosk.). 2010;44:1010-7
10. Zhao G, Cai C, Yang T. et al. MicroRNA-221 induces cell survival and cisplatin resistance through PI3K/Akt pathway in human osteosarcoma. PloS one. 2013;8:e53906
11. Codogno P. Shining light on autophagy. Nature Rev Mol Cell Biol. 2014;15:153
12. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728-41
13. Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656-64
14. Li L, Wang L, Xiao R. et al. The invasion of tobacco mosaic virus RNA induces endoplasmic reticulum stress-related autophagy in HeLa cells. Biosci Rep. 2012;32:171-86
15. Ogata M, Hino S, Saito A. et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220-31
16. Rasheva VI, Domingos PM. Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis. 2009;14:996-1007
17. Qin L, Wang Z, Tao L. et al. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy. 2010;6:239-47
18. McCarthy N. Autophagy: Directed development. Nature Rev Cancer. 2014;14:74-5
19. White E. Deconvoluting the context-dependent role for autophagy in cancer. Nature Rev Cancer. 2012;12:401-10
20. Huang J, Liu K, Yu Y. et al. Targeting HMGB1-mediated autophagy as a novel therapeutic strategy for osteosarcoma. Autophagy. 2012;8:275-7
21. Harding HP, Zhang Y, Bertolotti A. et al. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897-904
22. Vandewynckel YP, Laukens D, Bogaerts E. et al. Modulation of the unfolded protein response impedes tumor cell adaptation to proteotoxic stress: a PERK for hepatocellular carcinoma therapy. Hepatol Int. 2015;9:93-104
23. Harding HP, Zhang Y, Zeng H. et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619-33
24. Huang G, Yao J, Zeng W. et al. ER stress disrupts Ca2+-signaling complexes and Ca2+ regulation in secretory and muscle cells from PERK-knockout mice. J Cell Sci. 2006;119:153-61
25. Panaretakis T, Kepp O, Brockmeier U. et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009;28:578-90
26. Divieti P, Lanske B, Kronenberg HM. et al. Conditionally immortalized murine osteoblasts lacking the type 1 PTH/PTHrP receptor. J Bone Miner Res. 1998;13:1835-45
27. Bobrovnikova-Marjon E, Grigoriadou C, Pytel D. et al. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene. 2010;29:3881-95
28. Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nature Rev Cancer. 2014;14:581-97
29. Desai SD, Reed RE, Babu S. et al. ISG15 deregulates autophagy in genotoxin-treated ataxia telangiectasia cells. J Biol Chem. 2013;288:2388-402
30. Cherra SJ 3rd, Kulich SM, Uechi G. et al. Regulation of the autophagy protein LC3 by phosphorylation. J Cell Biol. 2010;190:533-9
31. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274-93
32. Jung CH, Ro SH, Cao J. et al. mTOR regulation of autophagy. FEBS letters. 2010;584:1287-95
33. Shigemitsu K, Tsujishita Y, Hara K. et al. Regulation of translational effectors by amino acid and mammalian target of rapamycin signaling pathways. Possible involvement of autophagy in cultured hepatoma cells. J Biol Chem. 1999;274:1058-65
34. Paglin S, Lee NY, Nakar C. et al. Rapamycin-sensitive pathway regulates mitochondrial membrane potential, autophagy, and survival in irradiated MCF-7 cells. Cancer Res. 2005;65:11061-70
35. Cullinan SB, Zhang D, Hannink M. et al. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198-209
36. Blais JD, Addison CL, Edge R. et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol Cell Biol. 2006;26:9517-32
37. Wang Y, Alam GN, Ning Y. et al. The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res. 2012;72:5396-406
38. Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis. Nature Rev Cancer. 2002;2:277-88
39. Atkins C, Liu Q, Minthorn E. et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013;73:1993-2002
Author contact
![]() Corresponding author: Prof. Guang-rong Ji, Department of Orthopaedics, The 2nd Affiliated Hospital of Harbin Medical University, No. 246, Rd Xuefu, District Nangang, Harbin, 150001, China. Tel: +86-451-8660-5152; Fax: +86-451-8660-5152; Email: jiguangrongsina.com or Jiguangrong08com.
Corresponding author: Prof. Guang-rong Ji, Department of Orthopaedics, The 2nd Affiliated Hospital of Harbin Medical University, No. 246, Rd Xuefu, District Nangang, Harbin, 150001, China. Tel: +86-451-8660-5152; Fax: +86-451-8660-5152; Email: jiguangrongsina.com or Jiguangrong08com.