Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(9):1026-1035. doi:10.7150/ijbs.12443 This issue Cite
Research Paper
DNA-PKcs Negatively Regulates Cyclin B1 Protein Stability through Facilitating Its Ubiquitination Mediated by Cdh1-APC/C Pathway
Zeng-Fu Shang1,2*, Wei Tan2,3*, Xiao-Dan Liu2*, Lan Yu4, Bing Li2, Ming Li1, Man Song1,2, Yu Wang2, Bei-Bei Xiao1, Cai-Gao Zhong3, Hua Guan2 ![]() , Ping-Kun Zhou1,2
, Ping-Kun Zhou1,2 ![]()
1. School of Radiation Medicine and Protection, Medical College of Soochow University; Collaborative Innovation Center of Radiation Medicine of Jiangsu Higher Education Institutions, Suzhou, Jiangsu 215123, P. R. China
2. Department of Radiation Toxicology and Oncology, Beijing Key Laboratory for Radiobiology (BKLRB), Beijing Institute of Radiation Medicine, Beijing 100850, P. R. China
3. School of Public Heath, Central South University, Changsha, Hunan Province, Changsha, Hunan 410078, P. R. China
4. Division of Molecular Radiation Biology, Department of Radiation Oncology, University of Texas Southwestern Medical Center at Dallas, Dallas, Texas 75390, USA
*These authors contributed equally to this work
Received 2015-4-19; Accepted 2015-5-24; Published 2015-7-14
Abstract
The catalytic subunit of DNA-dependent protein kinase (DNA-PKcs) is a critical component of the non-homologous end-joining pathway of DNA double-stranded break repair. DNA-PKcs has also been shown recently functioning in mitotic regulation. Here, we report that DNA-PKcs negatively regulates the stability of Cyclin B1 protein through facilitating its ubiquitination mediated by Cdh1 / E 3 ubiquitin ligase APC/C pathway. Loss of DNA-PKcs causes abnormal accumulation of Cyclin B1 protein. Cyclin B1 degradation is delayed in DNA-PKcs-deficient cells as result of attenuated ubiquitination. The impact of DNA-PKcs on Cyclin B1 stability relies on its kinase activity. Our study further reveals that DNA-PKcs interacts with APC/C core component APC2 and its co-activator Cdh1. The destruction of Cdh1 is accelerated in the absence of DNA-PKcs. Moreover, overexpression of exogenous Cdh1 can reverse the increase of Cyclin B1 protein in DNA-PKcs-deficient cells. Thus, DNA-PKcs, in addition to its direct role in DNA damage repair, functions in mitotic progression at least partially through regulating the stability of Cyclin B1 protein.
Keywords: Cyclin B1, DNA-PKcs
Introduction
DNA-PKcs, the catalytic subunit of DNA dependent protein kinase (DNA-PK) complex, is a critical component functioning in the non-homologous end joining (NHEJ) pathway of DNA double strand break (DSB) repair [1]. In response to DSB, the DNA-PK regulator subunits Ku70/Ku80 immediately bind to the free DNA ends, followed by recruitment of its catalytic subunit, DNA-PKcs to the DSB ends. This assembly induces rapid phosphorylation DNA-PKcs at several serine and threonine residues which mediated by ATM and DNA-PKcs itself. Among these phosphorylation sites, pT2609 and pS2056 phosphorylation cluster are clearly identified in vivo and have been proved to contribute to its DNA repair function. In addition to its role in DNA DSB repair, DNA-PKcs also involves in V(D)J recombination of immunoglobulin genes and T-cell receptor genes[2], and telomere length maintenance[3]. More interestingly, Lee et al and our group recently found that the phosphorylated DNA-PKcs/T2609 and S2056 localized at the centrosomes and spindle apparatus, and suppression of DNA-PKcs led to dysfunction of mitotic transition and terminally resulted in chromosomal instability[4, 5]. An increased phosphorylation of DNA-PKcs was also observed in the mitotic cells even without induction of DNA damage [4-7], while the exact molecular mechanism and potential substrates of DNA-PKcs functioning in mitosis have not been fully elucidated. A number of studies have demonstrated an increased level of Cyclin B1 protein in the cells when DNA-PKcs was depressed or inactivated [5, 8-10], suggesting that Cyclin B1 could be an important functional partner of DNA-PKcs in cell cycle regulation. It is well known that not only Cyclin B1 is obligated for the mitotic transition from G2 phase, but degradation of Cyclin B1 is also required for mitotic exit. Therefore, it is really important to elucidate whether and how DNA-PKcs affects Cyclin B1 protein expression.
Progression through the mitosis relies on the periodic fluctuation of the activity of Cdk1/cyclin B1 complex, also called maturation-promoting factor (MPF) [11]. Cyclin B1, the positive regulator of Cdk1/Cdc2 kinase, is upregulated during G2 phase due to the increased promoter activity and mRNA stability[12, 13]. Accumulation of Cyclin B1 induces formation of the Cdk1/Cyclin B1 complex. Binding of Cyclin B1 with the inactive phophorylatedCdk1 leads to conformational changes of Cdk1, and consequently alters its phosphorylation status and facilitates Cdk1 activity[14]. Once activated, Cdk1/Cyclin B1 complex can phosphorylate a series of substrates, which are crucial to the initiation of the chromosome condensation, nuclear envelope breakdown, and mitotic spindle assembly[15]. Activation of Cdk1/Cyclin B1 is essential for the transition of cells from G2 phase to mitotic phase. However, degradation of Cyclin B1 and inactivation of Cdk1 kinase is required for the cytokinesis and mitotic exit. Non-degradable Cyclin B1 leads to the phenotype of dose-dependent mitotic arrest [16, 17]. At the onset of anaphase, the degradation of Cyclin B1 through ubiquitin-mediated proteolysis is indispensable for accurate sister chromatid separation and chromosomal stability maintaining [18]. The E3 ubiquitin ligase anaphase promoting complex/ cyclosome (APC/C) was reportd to be responsible for poly-ubiquitinylating Cyclin B1 for destruction [19].
APC/C belongs to the RING finger family of ubiquitin E3 ligases, which is composed with many different subunits (APC1-8, APC9-11, and CDC26) and plays a crucial role in coordinating mitosis progression through targeting numerous regulators for destruction by the 26S proteasome [20, 21]. The recognition of APC/C with its substrates is thought to be mediated by its co-activators, the WD40 proteins Cdc20 (cell division cycle 20) and Cdh1(Cdc20 homologue 1) [22]. Cdc20 is activated in metaphase as soon as spindle checkpoint is satisfied, whereas Cdh1 is response for late mitosis and G1 phase events. During mitosis, the activity of APC/C is strictly controlled by spindle checkpoint to ensure that anaphase is only triggered after all sister chromatids attach properly to spindle microtubule [22]. Moreover, APC/C activity is also regulated through phosphorylation by Cdk1 and other mitotic kinases [23]. APC/C specific substrates, including cyclins A, B, PLK1, and Aurora kinases-A, are frequently upregulated in human tumors and are correlated with chromosomal instability and poor prognosis in cancers [21, 24, 25]. More importantly, in mice, Cdc20 mutant which losses the Mad2-binding function results in the development of spontaneous neoplasms, whereas Cdh1 heterozygosity develops epithelial tumors, suggesting APC/C play an essential role in suppressing tumorgenesis through maintaining chromosome stability [26, 27].
In this study, we demonstrate that DNA-PKcs negatively regulates Cyclin B1 protein stability through impacting the ubiquitination-mediated its proteolytic degradation. Both siRNA and DNA-PKcs specific inhibitor Nu7026 can attenuate Cyclin B1 ubiquitination. Furthermore, we find that DNA-PKcs depletion leads to deregulation of APC/C co-activator Cdh1. These results indicate that DNA-PKcs functions in mitosis through controlling the pathway of APC/C /Cdh1-mediated degradation of Cyclin B1 protein. This mitosis regulation function of DNA-PKcs contributes it another mechanistic way for maintaining genetic / chromosomal stability.
Materials and Methods
Cell lines and treatment
HeLa cells was maintained in DMEM (HyClone) supplemented with 10% fetal bovine serum (HyClone), 100 unites per ml penicilin and 100 μg/ml in a humidified incubator at 37°C with 5% CO2. DNA-PKcs activity inhibitor Nu7026 was purchased from Sigma, its stock solution was prepared in DMSO. Two hours before releasing from nocodazole (Sigma) block or CHX(Sigma) treatment, cells were pretreated with 10 μM Nu7026 or solvent concentration of DMSO and kept at this concentration during the subsequence treatment. Cells were synchronized at M phase by treated with nocodazole at 100 ng/ml concentration for 16h. After removal of the nocodazole, cells were cultured in fresh growth DMEM and harvested at different time points for cell cycle and western blotting analyses.
Antibodies
Cyclin B1 (GNS1), β-Actin (C4), p55 CDC (H-175), Cdc27 (AF3.1), ANAPC2 (H-295), Ub (P4D1), and DNA-PKCS(H-163) were purchased from Santa Cruz. Chd1 Ab-1(DH01) was purchased from Neo Markers. EasyBlot anti Rabbit and Mouse IgG (HRP) was purchased from GeneTex Irvine, CA.
Flow cytometric analysis of the cell cycle
After washing twice with PBS solution, cells were collected and fixed using 70% ethanol at -20ºC for at least 24 h. Then the cells were washed twice with PBS solution, incubated with 20 μl RNase A (1 mg/ml) for 30 min at 37ºC, and stained with 25 μg/ml PI for 30 min at room temperature. The cell cycle distribution was then analyzed using flow cytometry and more than 10,000 cells per sample were counted.
Immunoprecipitation and GST pull-down assays
For immunoprecipitation assay, cells were lysed with the lysis buffer [50 mmol/L Tris-HCl (pH7.5); 150 mmol/L NaCl; 0.2% Nonidet P-40; 5% glycerol; 1% (v/v) Tween 20; protease inhibitor cocktail (Roche)] and centrifuged at 12000 g for 10 min at 4ºC. Cells lysates were incubated with anti-Cyclin B1 antibody for 3 hours at 4ºC and then added protein A/G Sepharose (Santa Cruz) for overnight. The second day the Sepharose beads were washed with lysis buffer for three times and resuspended in SDS-PAGE loading buffer for western blotting analysis using corresponding antibodies. The GST fused Cyclin B1 protein were expressed in E. coli BL21 (DE3) cells grown at 37°C in 1 L of LB media to an optical density at 600 nm of 0.5 and then induced with 0.1 mM isopropyl-D-thiogalactopyranoside (IPTG) for 6 h at 25°C. Cells were washed twice with phosphate buffered saline solution (PBS). Cells were lysed on ice by sonication. The expressions of GST and GST-fusion proteins were purified by GST-Sepharose beads (Pharmacia) according to manufacturer's guidelines. HeLa cell lysates were incubated with the immobilized GST or GST-fusion protein at 4°C for 3 h. The beads washed with lysis buffer for 4 times and resuspended in SDS-PAGE loading buffer for western blotting analysis using anti-DNA-PKcs antibodies.
Ubiquitination assay
Cells were pretreated with DMSO or 10 μM Nu7026 for 2 hours and incubated with 10 μM MG-132 for 6 hours. After incubation cells were lysed and immunoprecipitated with anti-Cyclin B1 antibody as described above. Anti-Ub antibody was used to detect the ubiquitination.
RNAi assay
Cells were transfected with siRNA oligonucleotide using lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. 24 hours after the first transfection, a second transfection was performed. 24 hours after the second transfection cells were ready to use. The following siRNA target sequences were used: DNA-PKcs, 5' GGGCGCUAAUCGUACUGAATT 3'
Results
Suppression of DNA-PKcs increased the stability of the Cyclin B1 protein
Previous studies have revealed that DNA-PKcs was phosphorylated and activated during mitosis [5]. To identify its association with these well-described mitotic regulation proteins, DNA-PKcs was knocked-down using specific siRNA (DPKcs-siRNA), and the expression changes of mitosis regulated proteins were detected by western blotting. Cyclin B1 was found overexpressed in DNA-PKcs suppressed cells (DPKcs-siRNA) as compared the control cells which were transfected with the native control siRNA (NC-siRNA) (Figure 1A). To investigate whether the kinase activity of DNA-PKcs affects the expression of Cyclin B1 protein, we treated HeLa cells with 10µM DNA-PKcs specific inhibitor Nu7026 for 6h. As shown in Figure 1B, Nu7026 also increased the level of Cyclin B1 protein. This experiment was performed under the prerequisite of confirming the inhibition of Nu7026 on DNA-PKcs autophosphorylation at S2056.
DNA-PKcs deficiency increases the stability of Cyclin B1 protein. (A) Increased level of Cyclin B1 protein in DNA-PKcs-depressed HeLa cells by specific siRNA (DPK-siRNA). (B) DNA-PKcs inhibitor NU7026 increases Cyclin B1 expression. (C) Comparison of the degradation dynamics of Cyclin B1 protein between DNA-PKcs-depressed cells mediated by siRNA (DPK-siRNA) and control cells (NC-siRNA). Cells were treated with cycloheximide (CHX) for the indicated times to inhibit novel protein synthesis, and cell extracts were prepared followed by immunoblotting analysis for Cyclin B1. (D) Quantification of Cyclin B1 levels following CHX treatment based on densitometric scanning of the immunoblotting signals of the Cyclin B1 protein shown in Fig 1C. (E) Effect of DNA-PKcs inactivation by NU7012 on the stability of Cyclin B1 protein. After pretreated with NU7026 to inhibit DNA-PKcs, HeLa cells were incubated with cycloheximide (CHX) for the indicated times to inhibit novel protein synthesis, and the cell extracts were prepared followed by immunoblotting analysis of Cyclin B1.
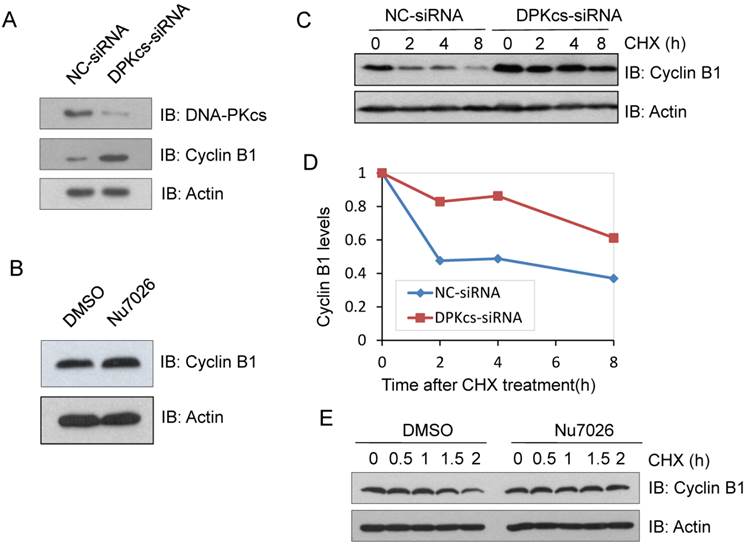
Cyclin B1 was reported to be mainly regulated at transcription level during G2/M transition and goes through proteasome degradation pathway at oneset of anaphase. The mRNA level of Cyclin B1 was determined by qRT-PCR in DNA-PKcs knockdown and control cells. On the contrary, the mRNA level of Cyclin B1 significantly decreased in DNA-PKcs deficient HeLa cells (data not shown), suggesting that the upregulation of Cyclin B1 in DNA-PKcs deficient cells may be a result of decreased protein degradation. To prove this hypothesis, the siRNA targeting-DNA-PKcs and negative control siRNA were transiently transfected into HeLa cells, 48h later the cells were treated with protein synthesis inhibitor cycloheximide (CHX) and harvested at different times after treatment. Immunoblotting was performed to detect the stability of Cyclin B1 protein. As shown in Figure 1C and D, the degradation of Cyclin B1 was significantly slower in the absence of DNA-PKcs. Moreover, inhibition of DNA-PKcs by Nu7026 also delayed Cyclin B1 degradation (Figure 1E), indicating that DNA-PKcs kinase activity is required for the regulation of Cyclin B1 stability.
Suppression of DNA-PKcs blocks Cyclin B1 ubiquitination
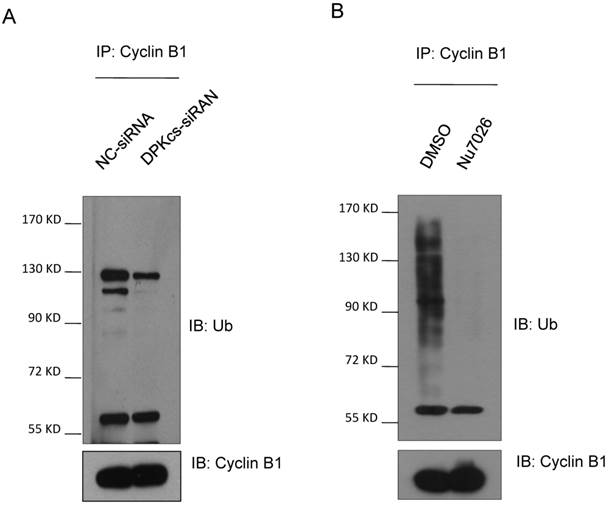
It has been suggested that Cyclin B1 is degraded through the ubiquitin/proteasome pathway mediated by APC/C complex. Based upon this evidence, we assumed that DNA-PKcs may participate in regulating Cyclin B1 ubiquitin/proteasome degradation pathway. Therefore, we firstly treated DNA-PKcs knockdown and control cells with proteasome inhibitor MG132 for 4h and then harvested the cells for immunoprecipitation experiments using anti-Cyclin B1 antibody. The immunoprecipitates were subject to western blotting experiments for determining whether DNA-PKcs has effect on the ubiquintination of Cyclin B1. As estimated, the polyubiquintinated Cyclin B1 in DNA-PKcs-depleted cells was dramatically decreased as compared to the control cells (Figure 2A). Moreover, DNA-PKcs inhibitor Nu7026 could also significantly inhibit Cyclin B1 polyubiquintination (Figure 2B), suggesting that the regulation effect of DNA-PKcs on Cyclin B1 ubiquintination is also dependent on its kinase activity.
Inactivation of DNA-PKcs attenuates the ubiquitination of Cyclin B1. (A) Depressing DNA-PKcs by specific DPK-siRNA attenuates the ubiquitination of cyclin B1 protein. Co-immunoprecipitated product of cyclin B1 was subjected to immunoblotting assay with anti-ubquitin antibody (upper panel), while 1/2 the amount of Cyclin B1 immunoprecipitates were subjected to immunoblotting analysis with Cyclin B1 antibody as a control (lower panel). (B) Inhibition of DNA-PKcs kinase activity by NU7026 decreases the ubiquitination of Cyclin B1 protein. HeLa cells were pretreated with DMSO or 10 μM Nu7026 for 2 hours and further incubated with 10 μM MG132 for 6 hours. The immunoprecipitation and immunoblotting assay were performed as indicated above.
The deregulation of APC/C co-activator Cdh1 is responsible for accumulation of Cyclin B1 in DNA-PKcs deficient cells
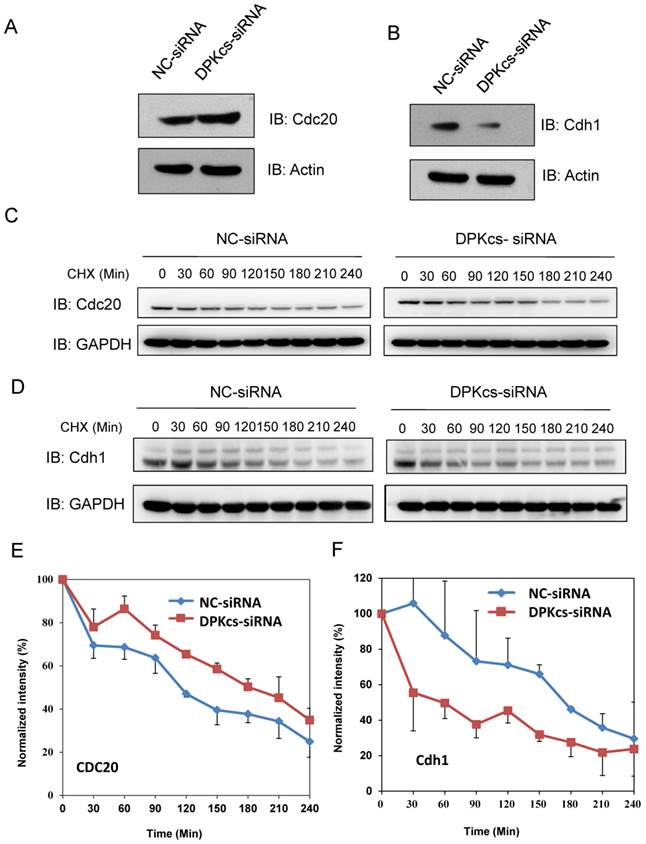
Cdc20 and Cdh1 are two co-activators, but functionally distinct, of E3 ubiquitin ligase APC/C. We further detect whether DNA-PKcs affects Cdc20 and Cdh1 levels. The western blotting analyses showed that DNA-PKcs deficiency increased slightly the level of Cdc20 protein (Figure 3A), while significantly decreased the level of cdh1 protein (Figure 3B). To investigate whether DNA-PKcs affects the protein stability, DNA-PKcs was transiently knocked-down by DNA-PKcs-targeting siRNA, and cycloheximide (CHX) was used to inhibit the de novo protein synthesis in HeLa cells. The result indicated that the half-life of Cdc20 was faintly prolonged (Figure 3C and E). Whereas, the Cdh1 degraded much faster in DNA-PKcs-deficiency cells when compared with control cells (Figure 3D and F).
The effects of DNA-PKcs deficiency on expression of APC/C co-activators Cdc20 and Cdh1. (A) Slightly increased expression of Cdc20 protein in DNA-PKcs-depressed HeLa cells by specific siRNA (DPK-siRNA). (B) Significantly decreased expression of Cdh1 protein in DNA-PKcs-depressed HeLa cells by specific siRNA (DPK-siRNA) as compared to control cells transfected with negative control siRNA (NC-siRNA). (C) Comparison of the degradation dynamics of Cdc20 protein between DNA-PKcs-depressed cells mediated by siRNA (DPK-siRNA) and control cells (NC-siRNA). Cells were treated with cycloheximide (CHX) for the indicated times to inhibit novel protein synthesis, and cell extracts were prepared followed by immunoblotting analysis for Cdc20. (D) Comparison of the degradation dynamics of Cdh1 protein between DNA-PKcs-depressed cells mediated by siRNA (DPK-siRNA) and control cells (NC-siRNA). The cells treatments and immunoblotting analysis were performed as above. (E) Quantification of Cdc20 levels following CHX treatment based on densitometric scanning of the Cdc20 immunoblotting signals as an example shown in Fig 4C. The data are the means and the standard deviation from three independent experiments. (F) Quantification of Cdh1 levels following CHX treatment based on densitometric scanning of the Cdh1 immunoblotting signals as an example shown in Fig 4D. The data are the means and the standard deviation from three independent experiments.
DNA-PKcs interacts with Cdh1 and Cyclin B1
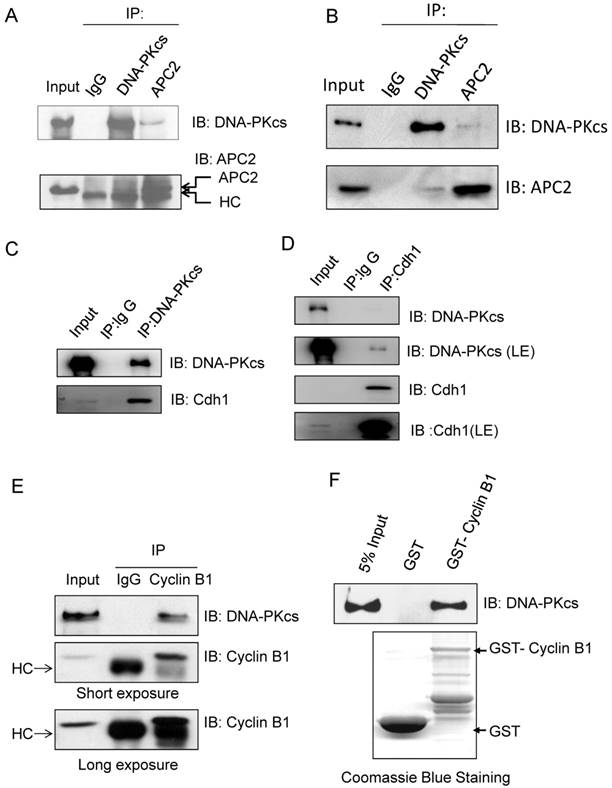
Our previous studies showed that DNA-PKcs is phosphorylated in mitotic phase, and the phosphorylated DNA-PKcs localizes at mitotic apparatus, including centrosomes, kinetochore and midbody during mitosis. Here we further demonstrated that DNA-PKcs regulates Cyline B1 stability via the APC/C pathway. We deduced that DNA-PKcs may interact with certain regulator(s) of Cyclin B1 in the same functional complex. To investigate this, immunoprecipitations were performed using anti-APC2, Cdh1, Cyclin B1 and DNA-PKcs antibodies. The results demonstrated the interaction of DNA-PKcs with APC/C core factor APC2 (Figure 4A) as well as Cdh1 (Figure 4C, D). The immunohybridizing bands of APC2 and IgG heavy chain (HC) are very close (Figure 4A) as the difference between both molecular weights is very small. We then repeated the experiment using the EasyBlot anti-rabbit IgG, which is an HRP-conjugated second step reagent that specifically reacts with the native, non-reduced form of mouse IgG and does not bind the reduced, denatured form IgG from the denatured Gel. As shown as Figure 4B, the iteraction of DNA-PKcs (DPKcs) and APC2 was displayed, and the non-specific signal disappeared. Therefore, DNA-PKcs might also play a role in APC/C-Cdh1 complex assembly. Moreover, DNA-PKcs and Cyclin B1 were also coimmunoprecipitated by the antibodies each other (Figure 4E). We further detected this interaction using GST-pull down assay, the result indicated that DNA-PKcs associated with GST-fused Cyclin B1, but not with GST (Figure 4F), indicating that DNA-PKcs can bind directly to Cyclin B1 in vitro.
Immunoprecipiation demonstrates the interaction of DNA-PKcs with APC/C co-activator and Cyclin B1. (A) DNA-PKcs interacts with APC2 in the cells' extract. Co-immunoprecipitation (CoIP) was performed using an antibody against DNA-PKcs or APC2, and the CoIP products were subjected to immunoblotting assay as indicated. (B) Repeat experiment on the interaction of DNA-PKcs with APC2. In order to eliminate the non-specific signal, the EasyBlot anti-rabbit IgG was used in this assay. This anti-IgG is an HRP-conjugated second step reagent that specifically reacts with the native, non-reduced form of mouse IgG and does not bind the reduced, denatured form IgG from the denatured Gel. (C, D) DNA-PKcs interacts with Cdh in the cells' extract. The Co-immunoprecipitation (CoIP) was performed using an antibody against DNA-PKcs (C) or Cdh1 (D), and the CoIP products were subjected to immunoblotting assay as indicated. (E) DNA-PKcs interacts with Cyclin B1 in the cells' extract. The Co-immunoprecipitation (CoIP) was performed using an antibody against Cyclin B1, and the CoIP products were subjected to immunoblotting assay of DNA-PKcs and Cyclin B1. (F) GST pull-down assay demonstrates the direct interaction of DNA-PKcs with GST-PLK1 in vitro and the lack of interaction with GST.
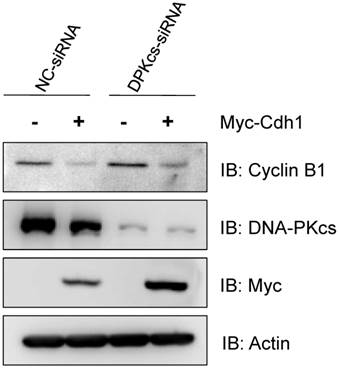
Overexpression of exogenous Cdh1 reverses the increase of Cyclin B1 protein caused by inactivation of DNA-PKcs
Finally, in order to see whether the depression of Cdh1 contributes to the accumulation of Cyclin B1, we transfected the exogenous Myc-tagged Cdh1 expressing vectors into the DNA-PKcs defective cells, and then detect the alteration of Cyclin B1 protein level. As shown in Figure 5, the increased level of Cyclin B1 protein was largely attenuated by overexpression of the exogenous Cdh1, suggesting that the accumulation of Cyclin B1 in DNA-PKcs deficient cells is associated with the depression of Cdh1 protein.
Overexpression of exogenous Cdh1 reverses the increase of Cyclin B1 protein in DNA-PKcs deficient cells. The cells were transfected myc-tagged Cdh1 expressing vectors, the cells extracts were subjected to immunoblotting analyses using the antibodies as indicated.
Discussion
Cyclin B1 is required for the mitotic entry, while the degradation of Cyclin B1 is also obligated for the mitotic exit after a cycling of cell. In this study, we revealed that DNA-PKcs is an upstream regulator of the Anaphase Promoting Complex (APC/C)-Cdh1 pathway mediating-Cyclin B1 degradation. We showed that DNA-PKcs deficiency attenuates the ubiquitination of Cyclin B1 and delays its destruction. Proteasome-dependent protein degradation depends on a special length of ubiquitin chain, while the formation of ubiquitin chain requires ubiquitination enzymes to retain its substrates during this progression. Our study demonstrated that DNA-PKcs facilitates the ubiquitination of Cyclin B1 mediated by APC/Ccdh1 E3 ligase, and suggesting that DNA-PKcs is a novel positive regulator of APC/C activity. As reported, APC/C core, especially APC3 is highly phosphorylated in mitosis, and the phosphorylation is crucial for the binding of Cyclin B1 with APC/C[23]. Both specific siRNA and chemical kinase inhibitor of DNA-PKcs can increase the half-life of Cyclin B1 protein, suggesting the regulation of DNA-PKcs in Cyclin B1 degradation was kinase activity dependent. Moreover, DNA-PKcs specific inhibitor Nu7026 could also suppress Cyclin B1 ubiquitination.
APC/C regulates cell cycle progression via formation of two closely related, but functionally distinct E3 ubiquitin ligase sub-complexes, APCCdc20 and APCCdh1, respectively. Cyclin B1 can interact with APC/C core or Cdc20 through its destruction box (D box) when the spindle checkpoint sacrificed. However, van Zon et al recently reported that the conserved Cdk cofactors Cks facilitate Cyclin B1 prior binding to APC/C Cdc20 through protein-protein interaction before spindle checkpoint released[23]. This previous binding mediated by Cks makes Cyclin B1 a better APC/C substrate. Our result demonstrated that DNA-PKcs not only physically interact with Cyclin B1, but also interact with the APC/C complex. These studies sheds light on another probable model in which DNA-PKcs may act as a scaffold and facilitate Cyclin B1 retention at the APC/C through its direct interaction with Cyclin B1. Further investigation will be needed to determine the exact acting mechanism of DNA-PKcs in APC/C complex assembly.
DNA-PKcs deficiency led to abnormal upregulation of Cyclin B1 protein. Our investigation suggested that DNA-PKcs regulates Cyclin B1 ubiquitination and degradation through regulating Cdh1 stability. Cdh1 is an important co-activator of APC/C complex, and there are multiple mechanistic pathways to regulate Cdh1 activity. Cdks-mediated phosphorylation of Cdh1 blocks its association with APC/C during S, G2 and mitosis, and during mitotic exit the binding of Cdh1 with APC/C core complex relies on inactivation of Cdks and dephosphorylation of Cdh1[20]. Except the regulation via post-translation modification, the amount of Cdh1 protein expression is also tightly controlled in a cell cycle dependent manner. Cdh1 can go through self-ubiquitination and degradation. Fukushima and co-workers' study has revealed that E3 ubiquitin ligase 3 SCF (β-TrCP) regulates APC/C-Cdh1 activity via promoting Cdh1 ubiquitination and degradation. In this process, PLK1 and Cyclin A/Cdk2 mediated phosphorylation of Cdh1 plays an essential role to facilitate the recognization by SCF. It was previously reported that DNA-PKcs interacted with PLK1 in mitotic phase [28, 29], and DNA-PKcs-depletion led to the increase of PLK1 protein [8, 28]. Here we found again that DNA-PKcs interacts with Cdh1. Therefore, we assumed that the increase of PLK1 by inactivating DNA-PKcs might contribute partially to the accelerated degradation of Cdh1. Recent work has also proved that PLK1 phsophorylates DNA-PKcs at its Ser3205 sites [29]. It would be interesting to determine whether the PLK1-mediated phosphorylation of DNA-PKcs participates in APC/C-Cdh1 activity regulation.
APC/C-Cdh1 can target Cyclin B1 and many other cell cycle regulators (such as PLK1 and Aurora A, B) for destruction, which is important for mitotic exit progression[30]. The failure of ubiquitination and degradation pathway of these targets mediated by APC/C-Cdh1 results in not only disorder of cell cycle, but also genetic / chromosomal instability, with closely associated with carcinogenesis [31-33]. Loss of DNA-PKcs has been shown leading to the occurrence of aneuploid cells and chromosomal instability [4, 28, 34]. Moreover, downregulated activity of DNA-PKcs in peripheral blood lymphocytes is associated with chromosomal instability and cancer risk [35, 36]. Growing evidences suggested that Cyclin B1 is over-expressed in many tumors [25, 37]. Overexpression of Cyclin B1 was suggested to challenge chromosomal stability through impairing spindle checkpoint and inducing incomplete cytokinesis[18]. Consistent with these phenomenon, our previous studies showed that silencing DNA-PKcs with siRNA or inhibiting its activity with small molecule inhibitor resulted in abnormalities in mitosis, including arrest at metaphase, abnormal spindle formation, failure of cytokinesis[28] and increased outcome of aneuploid cells[5, 38]. Hence, our works here shed light on the novel function of DNA-PKcs, a well characterized DNA DSB repair kinase, in cell cycle control and maintaining chromosome stability through targeting Cyclin B1 degradation may during mitotic exit.
Not only in normal mitosis, Cyclin B1 protein stability is also well regulated in response to DNA damage agents. Gillis et al demonstrated that Cyclin B1 is degraded in response to DNA damage in a p21 dependent manner, which is necessary for G2 checkpoint maintenance[39]. DNA-PK is also involved in the IR-induced G2 checkpoint response in human mammary epithelial cells [40]. A DNA damage checkpoint of mitotic exit was also reported to correlate with the attenuated degradation of Cyclin B1[41]. Elucidation, whether DNA-PKcs contributes to maintenance of DNA damage induced G2 checkpoint through promoting Cyclin B1 degradation, should be followed up in future studies.
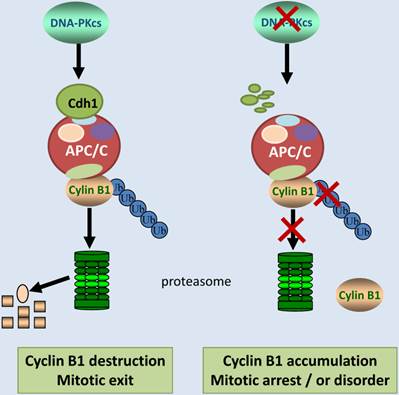
In summary, we report here that DNA-PKcs is an upstream regulator of Cyclin B1 ubiquitinaion and destruction medicated by APC/C-Cdh1 pathway. DNA-PKcs deficiency accelerated the degradation of Cdh1, and reduced Cdh1 protein level may lead to inactivation of E3 ubiquitin ligase 3 APC/C complex. As a result, the ubiquitination of cyclin B1 was attenuated, and consequently gave rise to Cyclin B1 accumulation (Figure 6). It is worthy to mention again, APC/C-Cdh1 is activated and functions in late mitosis, and destruction of Cyclin B1 is obligated for mitotic exit [42]. Inactivation of DNA-PKcs leads to accumulation of Cyclin B1, which may delay the mitotic exit or prolongs the mitotic arrest (Figure 6), consequently resulting in mitotic catastrophe or disorder as observed before in the DNA-PKcs defective cells upon to DNA damage[5]. Therefore, it would be interesting to further investigate whether DNA-PKcs plays a role in the mitotic exit checkpoint through controlling cyclin B1 destruction in response to DNA damage.
Schematic illustration shows the regulation pathway of DNA-PKcs on ubiquitination and degradation of Cyclin B1 protein. DNA-PKcs is required for the stability of Cdh1 protein, a co-activator of E3 ubiquitin ligase APC/C. Inactivation of DNA-PKcs accelerates Cdh1 degradation, resulting in the loss of APC/C activity. Therefore, Cyclin B1 protein can't undergo ubiquitination and destruction, consequently the accumulated Cyclin B1 could lead to mitotic arrest or exit delay.
Acknowledgements
This work was supported by National Key Basic Research Program of MOST, China (973 Program, grant #2015CB910601), the China National Science Foundation grant (#81472919), and Project Founded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhou PK. DNA Damage, Signaling and Repair: Protecting genomic integrity and reducing the risk of human disease. Chinese Science Bulletin. 2011;56:3119-21
2. Gellert M. V(D)J recombination: RAG proteins, repair factors, and regulation. Annu Rev Biochem. 2002;71:101-32
3. Bailey SM, Brenneman MA, Halbrook J, Nickoloff JA, Ullrich RL, Goodwin EH. The kinase activity of DNA-PK is required to protect mammalian telomeres. DNA Repair (Amst). 2004;3:225-33
4. Lee KJ, Lin YF, Chou HY, Yajima H, Fattah KR, Lee SC. et al. Involvement of DNA-dependent protein kinase in normal cell cycle progression through mitosis. The Journal of biological chemistry. 2011;286:12796-802
5. Shang ZF, Huang B, Xu QZ, Zhang SM, Fan R, Liu XD. et al. Inactivation of DNA-dependent protein kinase leads to spindle disruption and mitotic catastrophe with attenuated checkpoint protein 2 Phosphorylation in response to DNA damage. Cancer research. 2010;70:3657-66
6. Tu WZ, Li B, Huang B, Wang Y, Liu XD, Guan H. et al. gammaH2AX foci formation in the absence of DNA damage: mitotic H2AX phosphorylation is mediated by the DNA-PKcs/CHK2 pathway. FEBS Lett. 2013;587:3437-43
7. An J, Huang YC, Xu QZ, Zhou LJ, Shang ZF, Huang B. et al. DNA-PKcs plays a dominant role in the regulation of H2AX phosphorylation in response to DNA damage and cell cycle progression. BMC Mol Biol. 2010;11:18
8. Zhang B, Huang B, Guan H, Zhang SM, Xu QZ, He XP. et al. Proteomic profiling revealed the functional networks associated with mitotic catastrophe of HepG2 hepatoma cells induced by 6-bromine-5-hydroxy-4-methoxybenzaldehyde. Toxicology and applied pharmacology. 2011;252:307-17
9. Sun Y, Huang YC, Xu QZ, Wang HP, Bai B, Sui JL. et al. HIV-1 Tat depresses DNA-PK(CS) expression and DNA repair, and sensitizes cells to ionizing radiation. International journal of radiation oncology, biology, physics. 2006;65:842-50
10. Li B, Shang ZF, Yin JJ, Xu QZ, Liu XD, Wang Y. et al. PIG3 functions in DNA damage response through regulating DNA-PKcs homeostasis. International journal of biological sciences. 2013;9:425-34
11. Nurse P. Universal control mechanism regulating onset of M-phase. Nature. 1990;344:503-8
12. Hwang A, Maity A, McKenna WG, Muschel RJ. Cell cycle-dependent regulation of the cyclin B1 promoter. The Journal of biological chemistry. 1995;270:28419-24
13. Wang W, Caldwell MC, Lin S, Furneaux H, Gorospe M. HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO J. 2000;19:2340-50
14. Fattaey A, Booher RN. Myt1: a Wee1-type kinase that phosphorylates Cdc2 on residue Thr14. Prog Cell Cycle Res. 1997;3:233-40
15. Gavet O, Pines J. Activation of cyclin B1-Cdk1 synchronizes events in the nucleus and the cytoplasm at mitosis. J Cell Biol. 2010;189:247-59
16. Wolf F, Wandke C, Isenberg N, Geley S. Dose-dependent effects of stable cyclin B1 on progression through mitosis in human cells. EMBO J. 2006;25:2802-13
17. Chang DC, Xu N, Luo KQ. Degradation of cyclin B is required for the onset of anaphase in Mammalian cells. The Journal of biological chemistry. 2003;278:37865-73
18. Roh M, Song C, Kim J, Abdulkadir SA. Chromosomal instability induced by Pim-1 is passage-dependent and associated with dysregulation of cyclin B1. The Journal of biological chemistry. 2005;280:40568-77
19. van Leuken R, Clijsters L, Wolthuis R. To cell cycle, swing the APC/C. Biochimica et biophysica acta. 2008;1786:49-59
20. Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol. 2006;7:644-56
21. Kim HS, Vassilopoulos A, Wang RH, Lahusen T, Xiao Z, Xu X. et al. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer cell. 2011;20:487-99
22. Yu H. Cdc20: a WD40 activator for a cell cycle degradation machine. Molecular cell. 2007;27:3-16
23. van Zon W, Ogink J, ter Riet B, Medema RH, te Riele H, Wolthuis RM. The APC/C recruits cyclin B1-Cdk1-Cks in prometaphase before D box recognition to control mitotic exit. J Cell Biol. 2010;190:587-602
24. Song MS, Carracedo A, Salmena L, Song SJ, Egia A, Malumbres M. et al. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell. 2011;144:187-99
25. Qin L, Tong T, Song Y, Xue L, Fan F, Zhan Q. Aurora-A interacts with Cyclin B1 and enhances its stability. Cancer letters. 2009;275:77-85
26. Li M, Fang X, Wei Z, York JP, Zhang P. Loss of spindle assembly checkpoint-mediated inhibition of Cdc20 promotes tumorigenesis in mice. J Cell Biol. 2009;185:983-94
27. Garcia-Higuera I, Manchado E, Dubus P, Canamero M, Mendez J, Moreno S. et al. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat Cell Biol. 2008;10:802-11
28. Huang B, Shang ZF, Li B, Wang Y, Liu XD, Zhang SM. et al. DNA-PKcs associates with PLK1 and is involved in proper chromosome segregation and cytokinesis. Journal of cellular biochemistry. 2014;115:1077-88
29. Douglas P, Ye R, Trinkle-Mulcahy L, Neal JA, De Wever V, Morrice NA. et al. Polo-like kinase 1 (PLK1) and protein phosphatase 6 (PP6) regulate DNA-dependent protein kinase catalytic subunit (DNA-PKcs) phosphorylation in mitosis. Biosci Rep. 2014:34
30. Eguren M, Manchado E, Malumbres M. Non-mitotic functions of the Anaphase-Promoting Complex. Semin Cell Dev Biol. 2011;22:572-8
31. Zhang J, Wan L, Dai X, Sun Y, Wei W. Functional characterization of Anaphase Promoting Complex/Cyclosome (APC/C) E3 ubiquitin ligases in tumorigenesis. Biochimica et biophysica acta. 2014;1845:277-93
32. Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet. 2012;13:189-203
33. McGranahan N, Burrell RA, Endesfelder D, Novelli MR, Swanton C. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep. 2012;13:528-38
34. Hsu FM, Zhang S, Chen BP. Role of DNA-dependent protein kinase catalytic subunit in cancer development and treatment. Transl Cancer Res. 2012;1:22-34
35. Someya M, Sakata K, Matsumoto Y, Yamamoto H, Monobe M, Ikeda H. et al. The association of DNA-dependent protein kinase activity with chromosomal instability and risk of cancer. Carcinogenesis. 2006;27:117-22
36. Someya M, Sakata KI, Matsumoto Y, Kamdar RP, Kai M, Toyota M. et al. The association of DNA-dependent protein kinase activity of peripheral blood lymphocytes with prognosis of cancer. Br J Cancer. 2011;104:1724-9
37. Song Y, Zhao C, Dong L, Fu M, Xue L, Huang Z. et al. Overexpression of cyclin B1 in human esophageal squamous cell carcinoma cells induces tumor cell invasive growth and metastasis. Carcinogenesis. 2008;29:307-15
38. Shang Z, Yu L, Lin YF, Matsunaga S, Shen CY, Chen BP. DNA-PKcs activates the Chk2-Brca1 pathway during mitosis to ensure chromosomal stability. Oncogenesis. 2014;3:e85
39. Gillis LD, Leidal AM, Hill R, Lee PW. p21Cip1/WAF1 mediates cyclin B1 degradation in response to DNA damage. Cell cycle. 2009;8:253-6
40. Arlander SJ, Greene BT, Innes CL, Paules RS. DNA protein kinase-dependent G2 checkpoint revealed following knockdown of ataxia-telangiectasia mutated in human mammary epithelial cells. Cancer Res. 2008;68:89-97
41. Huang X, Tran T, Zhang L, Hatcher R, Zhang P. DNA damage-induced mitotic catastrophe is mediated by the Chk1-dependent mitotic exit DNA damage checkpoint. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:1065-70
42. Qiao X, Zhang L, Gamper AM, Fujita T, Wan Y. APC/C-Cdh1: from cell cycle to cellular differentiation and genomic integrity. Cell cycle. 2010;9:3904-12
Author contact
![]() Corresponding authors: Ping-Kun Zhou, E-mail: zhoupkac.cn; Hua Guan, Email: ghlshcom
Corresponding authors: Ping-Kun Zhou, E-mail: zhoupkac.cn; Hua Guan, Email: ghlshcom