Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusions
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(4):1497-1512. doi:10.7150/ijbs.102742 This issue Cite
Research Paper
Solobacterium moorei promotes tumor progression via the Integrin α2/β1-PI3K-AKT-mTOR-C-myc signaling pathway in colorectal cancer
Yan Chen1,2 ![]() , Ying Qin3, Tingting Fan4, Cheng Qiu2, Yijie Zhang1, Mengmeng Dai2, Yaoyao Zhou2, Qinsheng Sun1, Yuan Guo4, Yue Hao1
, Ying Qin3, Tingting Fan4, Cheng Qiu2, Yijie Zhang1, Mengmeng Dai2, Yaoyao Zhou2, Qinsheng Sun1, Yuan Guo4, Yue Hao1 ![]() , Yuyang Jiang1,2,5
, Yuyang Jiang1,2,5 ![]()
1. Guangdong Provincial Key Laboratory of Chinese Medicine Ingredients and Gut Microbiomics, School of Pharmacy, Shenzhen University Medical School, Shenzhen University, Shenzhen, Guangdong, China.
2. State Key Laboratory of Chemical Oncogenomics, Tsinghua Shenzhen International Graduate School, Shenzhen, Guangdong, China.
3. Department of Gastrointestinal Surgery, Shenzhen Second People's Hospital, Shenzhen, Guangdong, China.
4. Institute of Biomedical Health Technology and Engineering, Shenzhen Bay Laboratory, Shenzhen, Guangdong, China.
5. School of Pharmaceutical Sciences, Tsinghua University, Beijing, China.
Received 2024-8-23; Accepted 2025-1-7; Published 2025-1-27
Abstract

More and more evidences show that the imbalance of intestinal flora homeostasis can contribute to the progression of colorectal cancer (CRC). Solobacterium moorei (S. moorei), an anaerobic Gram-positive bacillus, was found to be enriched in fecal samples from CRC patients. However, the signaling regulatory mechanism of S. moorei promoting CRC progression remain unknown. Three CRC mouse models (ApcMin/+ mice, AOM/DSS-treated mice and subcutaneous colorectal xenograft mice) and two cell lines (DLD-1 and HT-29) were used to investigate the biological functions and molecular mechanisms of S. moorei on tumor progression of CRC in vivo and in vitro. S. moorei abundance increased in fecal samples and tumor tissues, and was significantly positively correlated with tumor staging of CRC. S. moorei promoted tumor progression in various CRC mouse models and it selectively adhered to cancer cells in comparison to colonic mucosal epithelial cells, enhancing CRC cell proliferation and inhibiting cell apoptosis. Mechanistically, S. moorei cellwall protein Cna B-type domain-containing protein binds to integrin α2/β1 on CRC cells, leading to the activation of PI3K-AKT-mTOR-C-myc pathway via phospho-FAK, thereby promoted tumor cell growth and progression. Blockade of integrin α2/β1 abolished S. moorei-mediated oncogenic response in vitro and in vivo. In summary, this study demonstrated that S. moorei promoted tumor progression via the integrin α2/β1-PI3K-AKT-mTOR-C-myc signaling pathway, which is a novel specific pathogen-mediated mechanism that might be a new potential target for CRC prevention, diagnosis, and treatment.
Keywords: Solobacterium moorei, Colorectal cancer, Tumor progression, Pathogen-mediated mechanism, Integrin α2/β1-PI3K-AKT-mTOR-C-myc signaling pathway
Introduction
Maintaining intestinal flora homeostasis is crucial for human health, and the imbalance of intestinal flora homeostasis leads to disease conditions[1, 2]. An increasing number of research studies have shown a strong link between intestinal flora and the progression and treatment of colorectal cancer (CRC) in humans[3-5]. Recently, the oral microbiome is recognized as a critical factor that affects intestinal flora homeostasis[6]. Oral microbiota is the second largest human microbiota after intestinal microbiota, and most oral microbial strains are delivered from the oral cavity to the large intestine[7]. Oral microbes have been increasingly recognized to be tightly associated with CRC[8, 9]. So far, oral bacteria such as Fusobacterium nucleatum, Porphyromonas gingivalis, Parvimonas micra and Peptostreptococcus anaerobius, were shown to invade human intestinal epithelium, which promoted the development of colorectal adenoma by several possible mechanisms[10-12].
Solobacterium moorei (S. moorei) is a gram-positive, non-spore-forming, anaerobic bacillus[13]. Unlike Fusobacterium nucleatum, Porphyromonas gingivalis and so on, S. moorei is an oral pathogen that has not been widely concerned. S. moorei was first identified in human fecal samples and described as a member of the oral microbiota[14]. It is a unique member of the genus Solobacterium in Clostridium cluster XVI. S. moorei should also be considered as an opportunistic pathogen, responsible for severe infections. S. moorei has been implicated in bacteraemia, necrobacillosis-associated thrombophlebitis, septicemia, and wound infection[15, 16]. S. moorei also has been detected in endodontic infections, periradicular lesions, and subgingival plaque of refractory periodontitis[17, 18]. In recent years, it has been found to be associated with oral malodour[19, 20] and oral squamous cell carcinoma[21]. Several microbiome sequencing studies reported that S. moorei is selectively enriched in the mucosal microbiota and fecal samples from CRC patients[22-24].
S. moorei has been reported to promote the progression of colon adenomas by causing inflammation and disrupting the intestinal barrier, demonstrating its important role in the process of inflammation-cancer transformation[25]. However, the relevant signal regulatory mechanisms are unknown. In addition, studies have shown that S. moorei exhibited a stronger replication rate during the adenoma phase, while its abundance significantly increased during tumor progression[23], indicating that in addition to playing a role in the early occurrence of CRC, S. moorei may also promote tumor development by regulating the proliferation of CRC cells. This suggests that S. moorei may become a potential candidate target for prevention and treatment of CRC. Therefore, in-depth research on the signaling pathways and key molecule mechanism involved in the regulation of tumor cell proliferation by S. moorei has great scientific and clinical value.
This study investigated the abundance of S. moorei in the feces and tumor of CRC patients and assessed its role in CRC development using mouse models and CRC cell lines. We found that S. moorei enhanced CRC occurrence by upregulating integrin α2/β1 expression in CRC cells, which is known to be overexpressed in human CRC. The interaction between integrin α2/β1 and S. moorei cellwall protein Cna B-type domain-containing protein activated the PI3K-Akt-mTOR pathway, resulting in C-myc activation and promoting tumor growth and progression. Our findings reveal a novel pathogen-mediated mechanism contributing to CRC progression.
Materials and methods
Analysis based on GMrepo database
GMrepo database was used to obtain data on microbial associations to host phenotypes for different species[26]. GMrepo (data repository for Gut Microbiota) is a database of curated and consistently annotated human gut metagenomes. The abundances of S. moorei in fecal samples of patients with CRC and healthy subjects were obtained from GMrepo and the relative abundance of S. moorei was analyzed. More detailed information is available on gmrepo.humangut.info/help.
Patients and clinical specimens
Clinical fecal specimens were respectively obtained from 96 healthy subjects and 89 CRC patients diagnosed by colonoscopy and biopsy from the health examination center and department of gastrointestinal surgery of Shenzhen Second People's Hospital. For healthy subjects, diarrhea was not reported in the past one month. Use of antibiotics and probiotics was not permitted from 1 month before the specimen collection. Freshly collected fecal specimens from CRC patients and healthy specimenwere frozen immediately after collection and preserved at -80 °C. The primary CRC tissues and the corresponding non-tumor tissues (5 cm away from the tumor edge) were obtained from patients who underwent surgery and were collected in RNAlater™ Stabilization Solution (Thermo Fisher Scientific, Waltham, MA, USA) and preserved at -80 °C refrigerator before analysis. The clinicopathological features of the samples are displayed in Table S1. CRC patients included in this study did not receive any treatment before surgery.
DNA and RNA extraction and qRT-PCR
QIAGEN stool kit (QIAGEN, Germany) was used to extract bacterial DNA from the fecal specimens of humans and mice, and QIAGEN DNA mini kit (QIAGEN, Germany) was used for extracting bacterial genomic DNA (gDNA) from human and mouse colonic samples. TRIzol reagent (Thermo Fisher Scientific) was used to isolate total RNA from cells and tissues. cDNA was synthesized by BeyoRT™ II thesis kit. RNA (1µg) was used for the reverse transcription reaction. Genomic DNA contamination was eliminated with gDNA Eraser kit (Beyotime Bio Inc, China). Thereafter, qRT-PCR assay was performed using a 7500 PCR system (Thermo Fisher Scientific). The PCR reaction conditions were as follows: initial denaturation at 95 °C for 5 min, followed by 40 cycles of denaturation at 95 °C for 10s, annealing and extension at 60 °C for 35s (Takara Bio, Japan). The relative abundance of S. moorei was calculated by the comparative CT method (2-ΔCt method in fecal specimens and 2-ΔΔCt method in tissue specimens). Universal eubacteria 16s rDNA was used as endogenous control. Further, the relative expression of the mRNA of the target genes was calculated by the 2-△Ct method, with GAPDH serving as a positive control. Primer sequences are enlisted in Table S2[11, 12, 27-31].
Bacterial Culture
S. moorei strain JCM10645 was purchased from the Japan Collection of Microorganisms (JCM, Japan). The hypervariable regions (V1-V9) of 16S ribosomal RNA (rRNA) were sequenced to confirm the identity of bacterial species. EG Broth Medium (BD Difco, Sparks, MD, USA) was used to grow bacteria in an anaerobic jar (80% N2, 10% CO2, and 10% H2) at 37 °C. Thereafter, cultures were collected, centrifuged at 3000 rpm for 5 min at 4 °C, washed twice with sterile anaerobic PBS, and resuspended at a concentration of 1 × 108 colony-forming units (CFU)/200 µL in an anaerobic environment for subsequent animal experiments. Meanwhile, E. coli strain MG1655 (ATCC 47076), a non-pathogenic human commensal gut bacterium, was used as a negative control (NC) for S. moorei infection. E. coli strain MG1655 was grown in lysogeny broth (LB) under aerobic conditions.
Colorectal adenoma mouse model
6-week-old C57BL/6 ApcMin/+ male mice (GemPharmatech, Jiangsu, China) and C57BL/6 wild type male mice (Medical laboratory animal center, Guangdong, China) were administered drinking water containing antibiotics (0.1g/L vancomycin, 0.2 g/L ampicillin, metronidazole, and neomycin) for 2 weeks. For the ApcMin/+ model, animals were randomized into three groups at one-week post-treatment, including mice receiving oral gavage of S. moorei (1 × 108 CFU), E. coli MG1655 (1 × 108 CFU), and PBS once every two days for 10 weeks. For the AOM/DSS-treated model, animals were randomized into four groups, (A) Normal control group: normal control with gavage daily PBS; (B) Model control group: AOM/DSS with gavage daily PBS; (C) E. coli group: AOM/DSS plus gavage 1 × 108 CFU E. coli suspension; (D) S. moorei group: AOM/DSS plus gavage 1 × 108 CFU S. moorei suspension. NC group received an injection of sterile saline, whereas the rest of the groups received an intraperitoneal injection of azoxymethane (AOM, Sigma-Aldrich, USA) solution at a doge of 10 mg/kg body weight at the beginning of the experiment. NC group was administered drinking water, whereas other groups were administered drinking water containing 2.5% dextran sulfate sodium salt (DSS, Yeasen, China) for 5 days followed by 14 days of normal water, this process repeated for three cycles. The bacteria were applied by oral gavage once every two days except when DSS was applied. After the completion of the experiment, mice were sacrificed, and the number and the volume of tumors or colon length in colon cancer mice were determined. Colon cancer tissues and matched non-tumor tissues were harvested, snap-frozen in liquid nitrogen, and preserved in 10% buffered formalin before use.
Subcutaneous colorectal xenograft mouse model
For the integrin α2/β1 blockade, DLD-1 cells (2 × 106) were inoculated into the left armpit of BALB/c-nu mice at 5 weeks of age. When the tumor grew to 100 mm3, the mice were randomly divided into four groups, receiving multipoint intratumoral injection of EG medium, S. moorei (1 × 108 CFU), RGD peptides (100 µg), and S. moorei plus RGD peptides once every two days for 2 weeks. The mice were euthanized before the tumors were dissected.
Histopathological analysis
Formalin-fixed paraffin-embedded tissues were used for histopathological analysis. 5 mm sections were prepared, stained with Haemotoxylin and Eosin (H&E), and used for histopathological analysis. H&E-stained sections were evaluated by experienced pathologists blinded to the treatments.
Cell Culture
Colon cancer cell lines DLD-1 and HT-29 were acquired from Cell Bank, China Academy of Sciences (Shanghai, China). Immortalized human colonic epithelial cell NCM460 was kindly provided by BeNa Culture Collection (Henan, China). DLD-1, HT-29, and NCM460 cells were cultured in RPMI-1640, McCoy's 5A, and DMEM (Gibco, Thermo Fisher Scientific), respectively. Each media contained 10% (v/v) fetal bovine serum (FBS), and the cells were maintained in a humidified incubator at 37 °C with 5% CO2. Each cell line was recently authenticated by STR profiling and tested for mycoplasma contamination. For the bacterial infection study, cells were infected with S. moorei anaerobically or E. coli MG1655 at MOI (multiplicity of infection) of 100 for 2 h every 24 hours. Following incubation, then removed the bacteria and the bacteria-containing medium was substituted by a cell culture medium that contained 10% FBS, 20 µg/mL gentamycin, and 1% penicillin-streptomycin (PS) continue to culture cells in the carbon dioxide incubator. Cells were co-cultured with bacteria three times within 72 hours.
Bacterial attachment assay
To study the attachment of S. moorei on colonic cells, NCM460, DLD-1, and HT-29 cells were co-cultured with 100 MOI for 2h in anaerobically condition, at that time S. moorei attached to cell surface(infected). After wash with PBS for 3 times, S. moorei which did not attach would be wash off. Then 10,000 cells attached with bacteria were plated in EG solid medium media which only allowed S. moorei to grow. Finally, we counted the formation of colony of S. moorei which represent the number of bacteria attached to cell surface. E. coli strain MG1655 was used as a negative control (NC). Each cell experiment was conducted thrice.
Transmission Electron Microscopy
Approximately 1 × 106 CRC cells were grown in a 6-well plate and co-cultured with S. moorei for 2h (MOI=10), then fixed in 2.5% glutaraldehyde. Fixed cell samples were further fixed using 1% osmium tetroxide, followed by dehydration with an increasing concentration gradient of ethanol and acetone. Samples were then embedded as followed: acetone: EMBed 812 epoxy = 1:1 for 2-4 h at 37°C; acetone: EMBed 812 epoxy = 1:2 overnight at 37°C; pure EMBed 812 epoxy for 5-8 h at 37°C. The resin blocks were cut to 60-80nm thin on the ultra-microtome, and the samples were fished out onto the 150 meshes cuprum grids with formvar film, then stained with 2% uranium acetate saturated alcohol solution and 2.6% lead citrate. The cuprum grids are observed under HITACHI TEM system and take images.
Cell Proliferation Assay
Nearly 5000 CRC cells were plated into a 96-well plate and co-cultured with S. moorei or E. coli MG1655 (MOI = 100) for 2 h every 24 hours, then removed the bacteria and continue to culture cells in the carbon dioxide incubator. CCK8 (Yeasen, China) kit was utilized to measure CRC cell viability in 24, 48, 72 hours after inoculation with S. moorei as described in the manufacturer's protocol. For the colony-forming assay, 1000 CRC cells were plated into a 24-well plate and also incubated with S. moorei or E. coli MG1655 (MOI = 100) for 2 h every 24 hours. After 72 hours, 1000 CRC cells were inoculated in a 6-well plate and cultured for 10 days. Following incubation, viable colonies (>50 cells/colony) were counted and stained with Giemsa. Each cell experiment was conducted thrice.
Cell apoptosis analysis
The cell apoptosis analysis was performed using flow cytometry. Cells were seeded into six-well plates and incubated with S. moorei or E. coli MG1655 (MOI = 100) for 2 h every 24 hours. After 72 hours, according to the manufacturer's instructions, apoptotic cells were measured with FITC AnnexinV Apoptosis Detection Kit (Yeasen, China).
RNA-sequencing analysis
Gene expression profiles of DLD-1 and HT-29 cells treated with or without S. moorei were analyzed by RNA sequencing. Nearly 1 × 106 colonic cells were infected with bacteria (MOI = 100) in a 6-well plate and maintained anaerobically for 4 hours. Cells were rinsed with PBS, and TRIzol reagent (Thermo Fisher Scientific) was utilized to extract total cellular RNA. RNA-seq was performed by BGI Technology Inc (Shenzhen, China). Gene Ontology (GO) analysis was performed to identify significantly enriched pathways using Metascape database[32] with the following criteria: the threshold of minimal count = 3, p-value < 0.01, and enrichment factor > 1.5.
Western blotting
Total cellular proteins were separated through SDS-PAGE and transferred on a PVDF membrane (BioRad, Hercules, CA). The membrane was incubated with primary and secondary antibodies sequentially (Table S3). The protein-antibody complex was detected using an enhanced chemiluminescence (ECL) detection system. Image analysis was performed to quantify protein expression levels with GAPDH as an internal control. Representative images of three independent experiments are shown in the Figures.
Knockdown of integrin α2/β1 within CRC cells
Lipofectamine 3000 Reagent (Invitrogen) was used for transfecting cells with small interfering RNAs (siRNA) specific for ITGA2 and ITGB1 (GenePharma, China) according to the manufacturer's protocol. Scrambled siRNA was used as a control. siRNA sequences utilized for inhibiting the expression ITGA2 and ITGB1 are displayed in Table S4. Cells transfected with siRNAs for 24 h were harvested for subsequent experiments.
Mass spectrometry identification of S. moorei proteins
1ⅹ109 amount of S. moorei were lysis and the protein were extracted through Gram-Positive Bacterium Protein Extraction Kit (Phygene, China). SDS-PAGE were performed and the protein gel were obtained for further LC/MS. The identification of protein gel strips is to separate the sample proteins by gel electrophoresis, then obtain the protein gel strips at different positions on the film, extract the peptides after enzymatic digestion. Separation was performed by Easy-nLC 1200 (Thermo Fisher Scientific, San Jose, CA). The nanoliter liquid phase separation end was directly connected to the mass spectrometer. The separated peptides were then ionized using a nanoESI source and analyzed by the Orbitrap Exploris 480 mass spectrometer (Thermo Fisher Scientific) in DDA mode. At last, the protein identification software which refer to Uniport database was used to identify the proteins in the samples.
Far-western assay
Whole-cell S. moorei proteins were extracted with Gram-Positive Bacterium Protein Extraction Kit (PH1710, Phygene), separated through SDS-PAGE and transferred on a PVDF membrane. The PVDF membrane was blocked with 5% BSA for 2h and incubated with human integrin α2/β1 heterodimer protein (ACRO Biosystems) overnight at 4 °C. The membrane was incubated with integrin α2/β1 and IgG primary antibody respectively for 12h at 4°C. After wash with TBST, membrane incubated with secondary antibody sequentially. The protein-antibody complex was detected using an ECL detection system.
Molecular docking for Integrinα2/β1 and Cna B-type domain-containing protein
Rigid protein-protein docking was performed between Integrinα2/β1 and Cna B-type domain-containing protein to investigate the relationships by using GRAMM-X (http://gramm.compbio.ku.edu/). The predicted protein structural domains of Integrinα2/β1 and Cna B-type domain-containing protein were obtained from the Alpha Fold 2 (https://alphafold.com/) and ITASSER (https://zhanggroup.org/I-TASSER/). Pymol (Version 2.4) and PDBePISA (https://www.ebi.ac.uk/pdbe/pisa/) were used to investigate protein-protein interactions and further visual analysis.
Expression and purification of Cna B-type domain-containing protein
Expression of Cna B-type domain-containing protein with His-tag was carried out in E. coli BL21(DE3) cells transformed with plasmids. Recombinant BL21(DE3) stored in glycerol was inoculated into TB medium containing related antibiotic and cultured at 37°C. When the OD600 reached about 1.2, cell culture was induced with 0.5mM IPTG at 15°C for 16h and was harvested by centrifugation. Recombinant BL21(DE3) were resuspended with lysis buffer followed by sonication. The precipitate after centrifugation was dissolved using denaturing agent. Target protein was obtained by two-step purification using Ni column+Q Sepharose column. Target protein after refolding was sterilized by 0.22 μm filter before stored in aliquots. The concentration was determined by Bradford protein assay with BSA as standard. The protein purity and molecular weight were determined by standard SDS-PAGE confirmation. Next, DLD-1 and HT-29 cells were treated with recombinant Cna B-type domain-containing protein at concentrations of 0, 0.05, 0.2, and 1 μg/ml, and the effects of the Cna B-type domain-containing protein on the proliferation and apoptosis of CRC cells were verified through CCK8 assay, colony-forming assay and flow cytometry.
Co-Immunoprecipitation
To validate interaction of Cna B-type domain-containing protein and integrin α2/β1, co-immunoprecipitation assay was performed. Briefly, 1ⅹ107 DLD-1 cells and 1ⅹ109 E. coli BL21(DE3) cells expressing Cna B-type domain-containing protein with His-tag were lysis and protein were extracted. Mixture of 200μL DLD-1 protein and E. coli protein were added and coupled to 50μL IgG Magnetic Beads (Beyotime, China) and Anti-His Magnetic Beads (Beyontime, China) for 24h, respectively. The beads are washed to remove unbound proteins, and the bound proteins are eluted with elution buffer. The eluted proteins are analyzed by Western blotting using specific antibodies to detect Cna B-type domain-containing protein with His-tag, integrin α2 and β1.
Statistical analysis
Statistical significance between the two groups was calculated by the student's t-test or Mann-Whitney U-test. One-way ANOVA was adopted to assess the statistical significance of cell growth curves. All in vitro experiments were repeated at least three times, results were represented by mean ± s.d. P < 0.05 (two-tailed) indicated statistical significance. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns: no significant.
Results
Solobacterium moorei enrichment in fecal and tumor specimens from CRC cases
We first analyzed S. moorei levels in the fecal samples of CRC cases and healthy subjects from GMrepo database. S. moorei levels were higher in CRC cases (n=390) in comparison to the healthy control samples (n=1589) (Figure 1A). To understand the association of S. moorei with CRC, the levels of S. moorei in the fecal specimens of 89 CRC cases and 96 healthy subjects were examined by qRT-PCR. S. moorei abundance was remarkably elevated in fecal specimens from CRC cases relative to healthy subjects and a significant association was observed between the S. moorei abundance in stools with the CRC tumor staging (Figure 1B). The abundance of S. moorei in the feces of stage III/IV CRC patients was significantly higher than that of stage I/II CRC patients. The abundance of S. moorei was also examined in tumor tissues and matched non-tumor tissues from CRC patients (Figure 1C). S. moorei abundance was remarkably elevated in tumor tissues and was also significantly correlated with tumor staging, suggesting the possible involvement of S. moorei in the progression of CRC.
The clinical relevance of S. moorei abundance with CRC. (A) The fecal abundance of S. moorei in the healthy controls (n =1589) and CRC patients (n =390) in GMrepo database. (B) The level of S. moorei in stool samples from healthy controls (n = 96), stage I/II CRC patients (n = 47) and stage III/IV CRC patients (n = 42). (C) The level of S. moorei in the tumor and non-carcinoma tissues from CRC patients (stage I/II, n = 47; stage III/IV, n = 42). HC, healthy control; CRC, colorectal cancer; SI/II, stage I/II; SIII/IV, stage III/IV.
S. moorei promotesd CRC occurrence among ApcMin/+ and AOM/DSS-treated C57BL/6 Mice
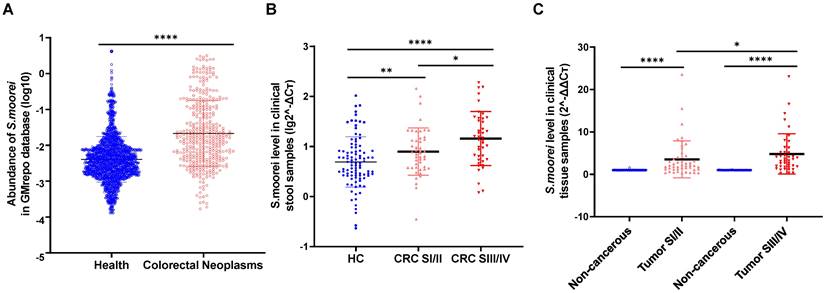
To assess the effect of S. moorei on CRC occurrence, the Spontaneous tumorigenesis model (ApcMin/+) and inducing tumorigenic model (AOM/DSS-treated) were utilized (Figure 2A-B). To deplete microbiota, antibiotics were administered to all mice for two weeks before the treatment. 1 × 108 colony-forming units (CFUs) of S. moorei were administered to each mouse via oral gavage in every 2 days in both ApcMin/+ and AOM+DSS models until the end of experiment. In addition, PBS or E. coli strain MG1655 (1 × 108 CFU) was administered to the mice as reference. qRT-PCR analysis of S. moorei in feces during feeding period confirmed that S. moorei colonized the intestinal tract of mice after inoculation (Figure S1A). The tumor development was examined at the end of experiment. Mice treated with S. moorei had markedly increased tumor multiplicity and tumor volume relative to those exposed to PBS or E. coli treatment (Figure 2C-F). Representative images of the colon from different treatment groups are displayed in Figure 2G-H. All colonic tumors were confirmed histologically, S. moorei strongly induced dysplasia in murine colon compared with those treated with E. coli MG1655 or PBS in both ApcMin/+ mice and AOM+DSS model (Figure 2I-J). qRT-PCR assay revealed an elevated level of S. moorei in colonic tumors compared to matched non-tumor samples (Figure S1B). Our data indicated that S. moorei accelerated colorectal tumorigenesis in both ApcMin/+ and AOM+DSS mice.
S. moorei promotes colonic tumorigenesis in ApcMin/+ and AOM/DSS-treated mice. (A) Schematic diagram showing the experimental design and timeline of ApcMin/+ mouse models. (B) Schematic diagram showing the experimental design and timeline of AOM/DSS-treated mouse models. (C) The colonic tumor number of ApcMin/+ mice under different treatments. (D) The tumor volume of ApcMin/+ mice under different treatment. (E) The colon length of AOM/DSS-treated mice under different treatment. NC, normal control; MC, model control. (F) The colonic tumor number of AOM/DSS-treated mice under different treatments. (G) Representative colonic morphologies of ApcMin/+ mice under different treatment. (H) Representative histological images of colon tissues of ApcMin/+ mice by H&E staining. Scale bars, 50 µm. (I) Representative colonic morphologies of AOM/DSS-treated mice under different treatment. NC, normal control; MC, model control. (J) Representative histological images of colon tissues of AOM/DSS-treated mice by H&E staining. Scale bars, 50 µm.
S. moorei attached preferentially to CRC cells
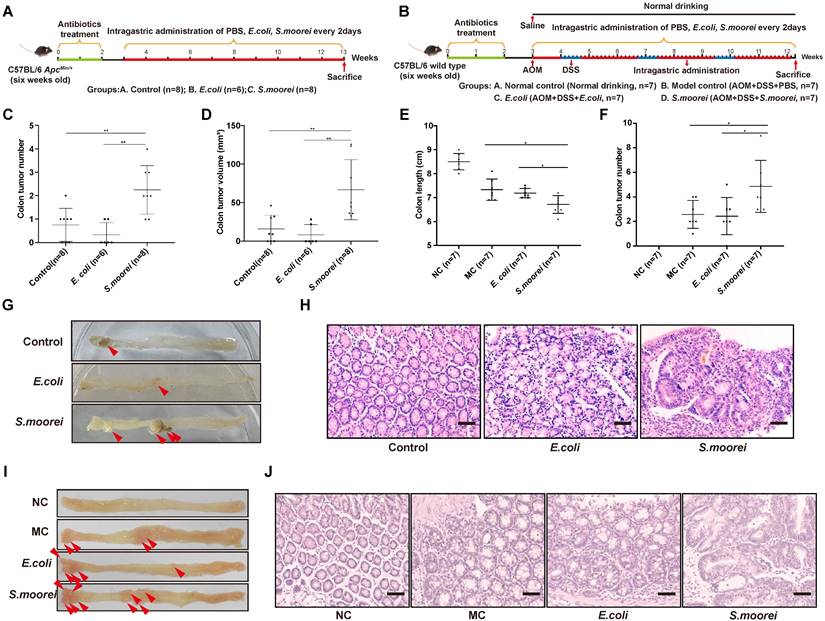
S. moorei were known to colonize in colonic tumors of ApcMin/+ mice; therefore, we analyzed in vitro adhesion characteristics of S. moorei to CRC cells (DLD-1 and HT-29) as well as human colonic mucosal epithelial cells (NCM460). We found that S. moorei successfully adhered to all the 3 cell types; however, the attachment efficiency of S. moorei was markedly elevated for CRC cells compared to primary epithelial cells (Figure 3A). On the contrary, E. coli strain MG1655 showed no obvious attachment to the three cell types (Figure 3B). Consequently, S. moorei possibly exhibited oncogenic activity by directly binding to colonic epithelial cells, especially CRC cells. Further, we also performed transmission electron microscopy (TEM) to confirm that S. moorei attaches to CRC cells (Figure 3C).
S. moorei attaches preferentially to CRC cells. (A) The level of attachment of S. moorei on colon cancer cell lines DLD-1 and HT-29 and the normal colon cell line NCM460. (B) The level of attachment of E. coli (MG1655) on colonic cells. (C) Representative TEM images of S. moorei attaching to colon cancer DLD-1 and HT-29 cells. The red arrows indicate S. moorei. Scale bars, 1 μm in S. moorei (The uppermost panel); 2 μm for the remaining panels.
S. moorei induced cell proliferation in vitro
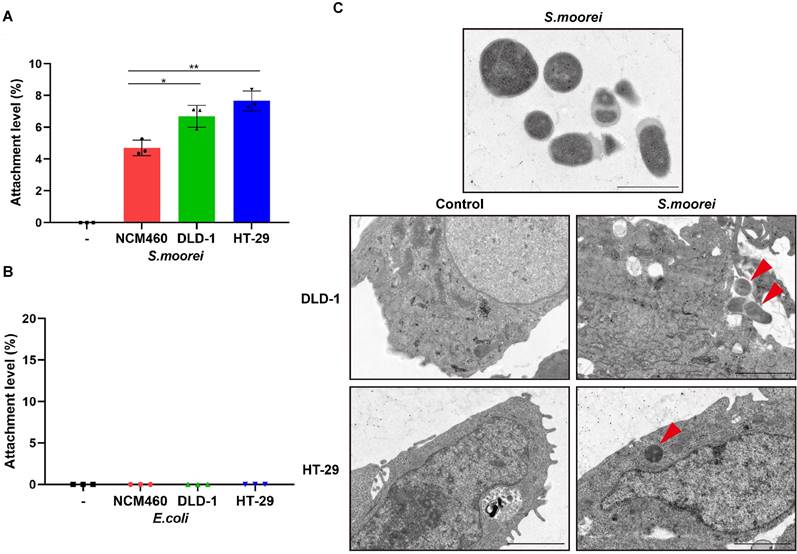
To further investigate the tumorigenic ability of S. moorei, the effect of S. moorei on cell proliferation was examined in vitro. Anaerobic culture of CRC cells for 2 hours had no effect on cell viability (Figure 4A). Additionally, S. moorei markedly enhanced the proliferation and the clone forming of DLD-1 and HT-29 cells, but the proliferation of CRC cells did not alter when co-cultured with the control bacteria E. coli (Figure 4B-E). Furthermore, Flow cytometry showed that S. moorei could inhibit the apoptosis of DLD-1 and HT-29 cells, while E. coli had no significant effect on cell apoptosis (Figure 4F-G). We also analyzed the expression of proteins involved in cell proliferation, cell cycle, and apoptosis in CRC cells following treatment with S. moorei, S. moorei increased the expression of PCNA, Cyclin D1, Bcl-2 and reduced Bax expression (Figure S2A-B). Taken together, our data suggested that S. moorei induced the tumorigenicity of CRC by accelerating cell proliferation and inhibiting the cell apoptosis.
S. moorei promotes cell proliferation and inhibits cell apoptosis in CRC cells. (A) Viabilities of CRC cells under anaerobic incubation condition for 2 hours were not altered. (B) Co-cultured with S. moorei promoted cell proliferation of DLD-1 but E. coli has no effect on cell viability. (C) S. moorei promoted cell proliferation of HT-29 cells. (D) Co-cultured with S. moorei promoted colony formation of DLD-1 and HT-29 cells but E. coli has no effect. (E) The colony formation number of DLD-1 and HT-29 treated with S. moorei. (F) Co-cultured with S. moorei inhibited cell apoptosis of DLD-1 but E. coli has no effect. (G) S. moorei inhibited cell apoptosis of HT-29 cells.
S. moorei promoted colorectal carcinogenesis by activating the integrin α2/β1-PI3K-AKT-mTOR-c-Myc signaling pathway
To understand the mechanism of S. moorei-induced CRC occurrence, gene expression levels in DLD-1 and HT-29 cells co-cultured with S. moorei were analyzed (Tables S5 and S6). To identify key cellular pathways activated after bacterial treatment, we utilized the Metascape database for GO as well as KEGG analysis. The enriched GO terms and KEGG pathways in DLD-1 and HT-29 cells following co-culture with S. moorei were identified (Tables S7 and S8). Based on the 50 most significantly enriched functions and pathways with p < 0.01, a Venn diagram of the significantly altered pathways common in two CRC cell lines was constructed. Overall, 9 significantly altered pathways in two CRC cell lines were identified, including the transmembrane receptor protein tyrosine kinase pathway, metabolism of nucleotides, response to oxygen level variations, and PI3K-Akt signaling pathway (Figure 5A). Integrins play an important role in apical junction remodeling by inducing the phosphorylation of focal adhesion kinase (FAK), which subsequently phosphorylate the p85 subunit of PI3K and activate the PI3K-AKT pathway[33, 34]. From the RNA-seq analysis, 356 genes (p < 0.05) were upregulated in CRC cell lines co-cultured with S. moorei (Table S9). Among those genes, ITGA2 and ITGB1 are the integrin family genes which activate the PI3K-AKT pathway in Peptostreptococcus anaerobius-mediated oncogenic response in CRC[12]. Meanwhile, MYC which is the downstream gene of PI3K-AKT pathway is also up-regulated. Therefore, we assumed that integrin α2/β1-PI3K-AKT-C-myc signaling pathway might be responsible for the tumor-promoting activity of S. moorei. For further validation, we analyzed the expression levels of genes involved in the PI3K-AKT pathway. We found that key genes related to the PI3K-AKT pathway were significantly increased in CRC cell lines co-cultured with S. moorei, including ITGA2, ITGB1, PTK2(FAK), PIK3R1, AKT1, MTOR and MYC (Figure 5B and S3A). Moreover, S. moorei induced the expression of several proteins, including Integrin α2/β1, p-FAK, p-PI3K (p-p85), p-AKT, p-mTOR and C-myc (Figure 5C-D and S3B-C). Our findings suggested that S. moorei promoted Integrin α2/β1-PI3K-AKT-mTOR-C-myc signaling pathway in CRC cells.
S. moorei mediates integrin α2/β1 signaling to activate the FAK-PI3K-AKT-mTOR-C-myc pathway to promote cell proliferation in vitro. (A) S. moorei regulated genes enrichment in numbers of pathways in DLD-1 and HT-29 cells. Nine pathways were commonly enriched in the two CRC cell lines. (B) Gene expression of integrin α2/β1-FAK-PI3K-AKT-mTOR-C-myc signaling pathway in DLD-1 cells co-cultured with S. moorei. (C) Protein expression of integrin α2/β1-FAK-PI3K-AKT-mTOR-C-myc signaling pathway triggered by S. moorei in DLD-1 cells. The effects of S. moorei were abolished by the integrin inhibitor RGD peptides (100 µM). RGDS, RGD peptides. (D) Quantification of protein expression for all blots showed in (C). (E) Integrin α2/β1 knockdown abolished the effect of S. moorei on the integrin α2/β1-FAK-PI3K-AKT-mTOR-C-myc pathway in DLD-1 cells. (F) Quantification of protein expression for all blots showed in (E).
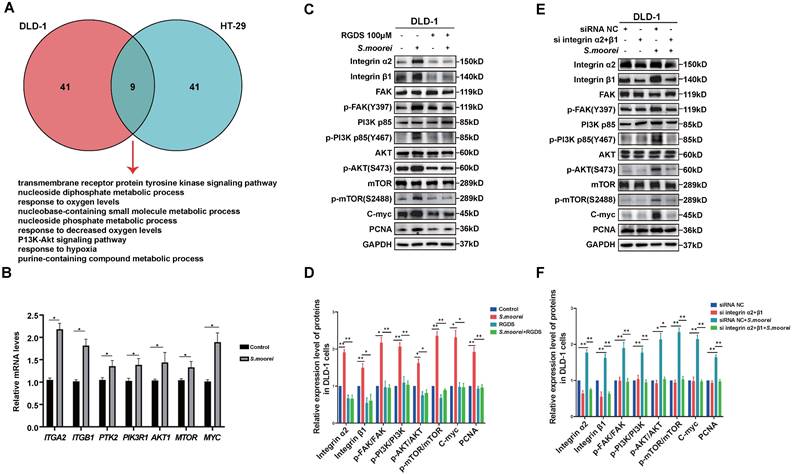
Blockade of integrin α2/β1 inhibited tumorigenic and signaling activities of S. moorei in CRC cells
To investigate the role of integrin α2/β1 signaling in S. moorei-mediated tumorigenic activity of CRC cells, the function of integrin α2/β1 was suppressed by integrin inhibitor RGD peptides or integrin α2/β1 small interfering RNA. Co-culture of CRC cells with S. moorei in the presence of RGD peptides abrogated the S. moorei-induced integrin α2/β1-PI3K-AKT-mTOR-C-myc signalling cascade and PCNA protein expression (Figure 5C-D and S3B-C). RGD peptides also blocked the increase in clone formation of DLD-1 and HT-29 cells induced by S. moorei while RGDS alone has no significant effect on clone formation. (Figure S3D-E). siRNA-mediated silencing of integrin α2/β1 in CRC cells also showed similar effects following incubation with S. moorei, integrin α2, integrin β1, p-FAK, p-PI3K, p-AKT, p-mTOR, C-myc, and PCNA protein levels were also downregulated in CRC cells exposed to S. moorei following transfection with siRNA targeting integrin α2/β1 (Figure 5E-F and S3F-G). The above findings corroborated that S. moorei induced integrin α2/β1-PI3K-AKT-mTOR-C-myc signaling pathway in CRC cells.
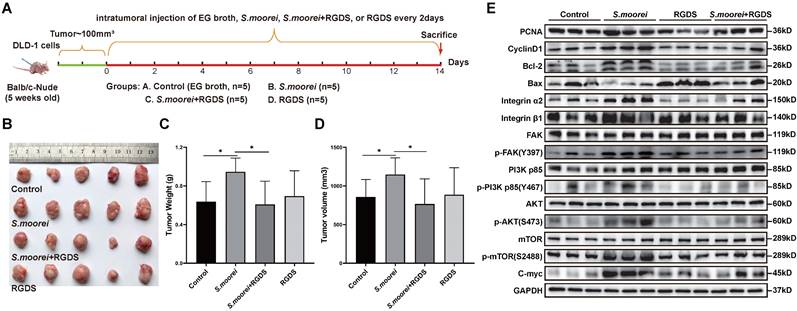
S. moorei activated the integrin α2/β1 signaling cascade and blockade of integrin α2/β1 inhibited tumorigenic and signaling activities of S. moorei in vivo
To investigate the in vivo effect of S. moorei in altering cell signaling pathways, qRT-PCR assay was conducted to analyze gene expression levels in colonic tumors of ApcMin/+ mice receiving S. moorei or vehicle treatment. Expression levels of Pcna, Ccnd1(CyclinD1), Bcl2, Itga2, Itgb1, Fak, Pik3r1, Akt1, Mtor and Myc were remarkably upregulated and Bax was downregulated in colonic tumors of mice treated with S. moorei (Figure S4A). Moreover, the expression of PCNA, CyclinD1, Bcl-2, Integrin α2/β1, p-FAK, p-PI3K (p85), p-AKT, p-mTOR, C-myc proteins was also significantly increased in the tumors of ApcMin/+ mice treated with S. moorei (Figure S4B). Then, the DLD-1 xenograft tumor model was used to investigate the role of S. moorei in promoting colorectal carcinogenesis and the effect of integrin α2/β1 signaling blockade on the tumorigenic activities of S. moorei. Mice with subcutaneous colorectal cancer were intratumorally injected with EG broth, S. moorei, RGD peptides, and S. moorei plus RGD peptides and the tumor tissue was harvested 2 weeks later (Figure 6A). The introduction of S. moorei into the subcutaneous xenografts in mice increased tumor weight and volume but RGD peptides treatment reversed the effect of S. moorei on the increase in tumor weight and volume (Figure 6B-D). The use of EG both or RGD peptides alone had no effect on tumor growth. In addition, protein expression of PCNA, CyclinD1, Bcl-2, Integrin α2/β1, p-FAK, p-PI3K (p85), p-AKT, p-mTOR, C-myc proteins were also significantly increased in the DLD-1 xenograft tumor treated with S. moorei and were reversed by RGD peptides in S. moorei-treated DLD-1 xenograft tumor (Figure 6E). These findings were consistent with in vitro results, indicating that S. moorei contributed to the activation of integrin α2/β1-PI3K-AKT-mTOR-C-myc signaling cascade.
S. moorei activates the integrin α2/β1 signaling cascade and blockade of integrin α2/β1 inhibited tumorigenic and signaling activities of S. moorei in DLD-1 xenograft tumor model. (A) Schematic diagram showing the experimental design and timeline of DLD-1 xenograft tumor model. (B) The images of DLD-1 xenograft tumor under different treatment. (C) The tumor weight under different treatment. (D) The tumor volume under different treatment. (E) Protein expression of proliferation/apoptosis-related genes and integrin α2/β1-FAK-PI3K-AKT-mTOR-C-myc signaling pathway in tumors of DLD-1 xenograft tumor model under different treatment. RGDS, RGD peptides.
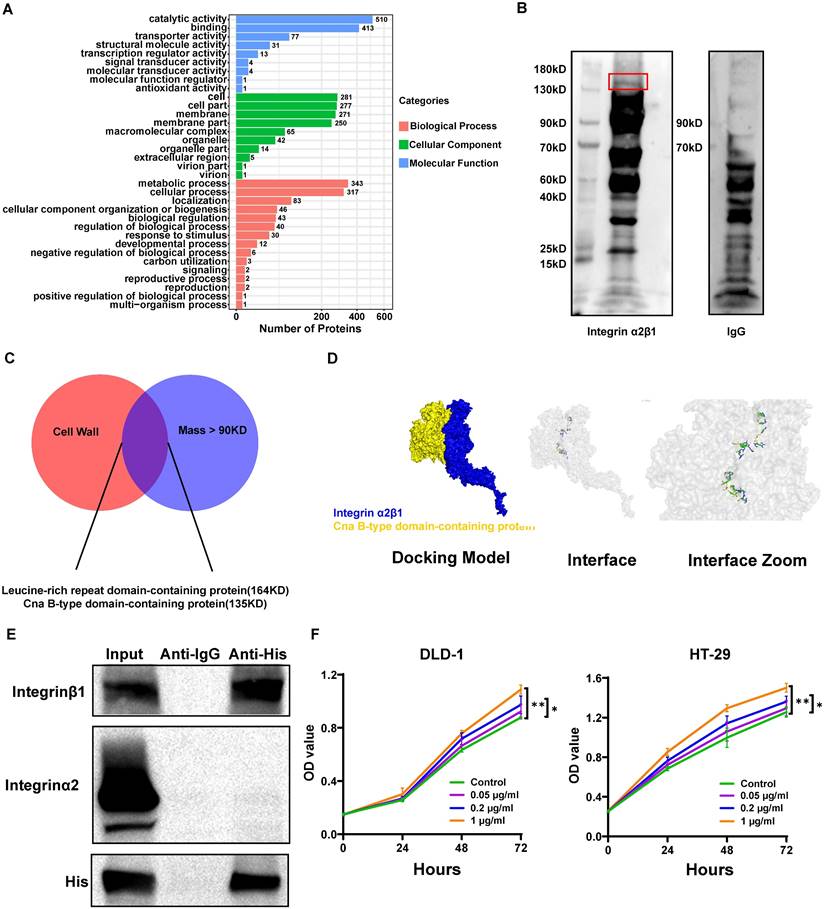
The S. moorei cellwall protein Cna B-type domain-containing protein binds to integrin α2/β1 on CRC cells
Since S. moorei attached preferentially to CRC cells and activates the integrin α2/β1- signaling pathway, we next to identify the surface proteins capable binding to the integrin α2/β1 of CRC cells. We first identified 805 proteins of S. moorei through mass spectrometry and clarified their localization (Table S10). Among the 805 proteins mentioned above, 10 proteins are located on the cellwall. Figure 7A shows the GO pathway enriched with S. moorei proteins. Using the far-western method, we identified the corresponding bands of protein from S. moorei that interacted with human integrin α2/β1 heterodimer protein. The results showed that compared with the IgG control group, the molecular mass of the S. moorei protein significantly bound to integrin α2/β1 was greater than 90 kDa (Figure 7B). Leucine-rich repeat domain-containing protein (164 kDa) and Cna B-type domain-containing protein (135 kDa) were S. moorei cellwall proteins which have a molecular mass greater than 90 kDa (Figure 7C). Considering that the protein band is closer to the molecular mass of 130 kDa, we speculate that Cna B-type domain-containing protein is the S. moorei cellwall protein binding to integrin α2/β1. To validate the binding of S. moorei Cna B-type domain-containing protein to integrin α2/β1 heterodimer protein, we performed rigid protein-protein docking between integrin α2/β1 and Cna B-type domain-containing protein. As shown in Figure 7D, integrin α2/β1 and Cna B-type domain-containing protein conjugated together through hydrogen bonds which formed by amino acid residue sites, revealing they formed a stable docking model (Supplementary Table S11). We subsequently constructed a plasmid encoding the Cna B-type domain-containing protein with a His tag and transfected it into E. coli BL21(DE3) cells for protein expression (Figures S5A-S5B). Results from the Co-IP experiments indicated that the Cna B-type domain-containing protein binds to integrin β1 in CRC cells (Figure 7E). Additionally, we assessed the impact of the recombinant Cna B-type domain-containing protein on CRC cell proliferation. The findings revealed that at concentrations of 0.2 and 1 μg/ml, the Cna B-type domain-containing protein significantly enhanced the proliferation and colony formation of DLD-1 and HT-29 cells (Figure 7F and S5C) while markedly inhibiting their apoptosis (Figure S5D). These results suggest that the Cna B-type domain-containing protein may play a crucial role in the oncogenic activity of S. moorei. Collectively, our findings validate the underlying role of the integrin α2/β1-PI3K-AKT-mTOR-C-myc signaling cascade in S. moorei-mediated colorectal tumorigenesis (Figure 8).
The S. moorei cellwall protein Cna B-type domain-containing protein binds to integrin α2/β1 on CRC cells. (A) Gene ontology for protein of S. moorei. (B) Far western blotting for protein of S. moorei interacted with integrinα2/β1. (C) Venn diagram for protein identified location on cell wall and mass over than 90KD. (D) Surface diagram of the docking model and their interfacing residues between Cna B-type domain-containing protein and integrinα2/β1. (E) Co-IP analysis demonstrating the interaction between the Cna B-type domain-containing protein (with His-tag) and integrin α2/β1, confirming the binding of the B-type domain-containing protein to integrin β1. (F) The effect of different concentrations of recombinant Cna B-type domain-containing protein on the proliferation of DLD-1 and HT-29 cells.
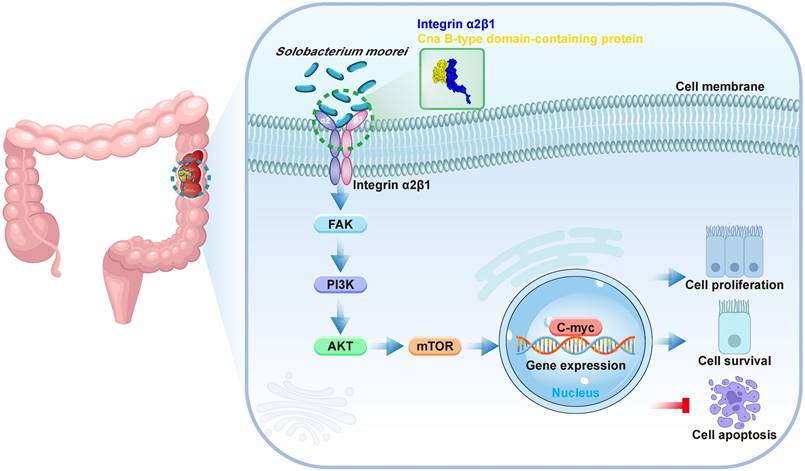
Schematic model depicting the proposed mechanism of S. moorei-mediated progression of colorectal cancer.
Discussion
Microbial dysbiosis has been increasingly suggested to facilitate the progression of CRC[35]. Metagenomic profiling was conducted on fecal and mucosal specimens collected from CRC patients, which suggested that S. moorei was one of the oncogenic bacteria that is highly enriched in CRC[22, 23]. The present study confirmed that S. moorei was enriched in CRC and was also significantly correlated with tumor staging. The present study confirmed that S. moorei was enriched in CRC by qRT-PCR analysis. S. moorei was enriched in fecal specimens of CRC patients in comparison to healthy subjects. Moreover, S. moorei was highly enriched in tumor tissues of CRC patients. Following microbiota depletion, S. moorei treatment dramatically accelerated the progression of CRC compared to the ApcMin/+ or AOM/DSS-treated mice exposed to E. coli strain MG1655. In addition, S. moorei promoted the growth of CRC cells. Collectively, our data suggested that S. moorei infection was linked to CRC and might be a candidate driver bacterium responsible for the progression of CRC. The mechanism of S. moorei-mediated progression of CRC is still unclear. The present work elucidated a mechanism by which S. moorei promoted cell proliferation in CRC. S. moorei particularly accumulated in the tumor tissue, indicating its possible interaction with CRC cells. Similar to in vivo findings, S. moorei showed attachment to CRC cells and colonic epithelial cells in vitro; however, the attachment efficiency of S. moorei was much higher for CRC cells compared to colonic epithelial cells. Notably, the attachment and colonization of cancer-promoting bacteria created a conducive environment for cancer progression, which was observed during F. nucleatum, B. fragilis, P. anaerobius, and P. gingivalis infections[12, 36-38]. We detected an altered gene expression profile in CRC cells after S. moorei treatment using transcriptome sequencing. Treatment with S. moorei induced the expression of ITGA2, ITGB1, and MYC genes in CRC cells. Gene enrichment analysis showed significant changes in the expression of genes involved in the PI3K-AKT pathway.
Integrins are the surface adhesion receptors mediating interactions between neighboring cells and the extracellular matrix[39]. Following binding to their ligands, integrins activate cell signaling pathways by recruiting and activating signaling proteins, including FAK[40]. FAK is responsible for the phosphorylation of different adaptor proteins, like PI3K, and subsequently activating the PI3K-AKT pathway[41]. Activated FAK-PI3K-AKT pathway accelerates cell motility, cell survival, and cell cycle progression[42]. FAK-PI3K-AKT pathway has been found to be activated in different cancer types and induces the proliferation and migration of tumor cells[43, 44]. It was reported that integrin α2/β1 complex was overexpressed in CRC cells compared to colonic epithelial cells[12], which might contribute to increased S. moorei attachment to tumor tissues. Additionally, S. moorei stimulated the expression of integrin α2/β1 levels in CRC cells, promoting cell adhesion. Likewise, pathogenic bacteria are shown to bind to integrins of CRC cells during pathogenesis. In tumor tissues from S. moorei-exposed ApcMin/+ mice and cultured CRC cells, integrin α2/β1 complex induced FAK phosphorylation which subsequently activated PI3K-AKT-mTOR-C-myc oncogenic signaling cascade. Indeed, S. moorei promoted cell growth in the ApcMin/+ and AOM/DSS-treated mice models and two CRC cell lines in vitro. RGD peptides or siRNA targeting integrin α2/β1 abolished S. moorei-mediated cell growth and the activation of the PI3K-AKT pathway, which suggested that the binding of bacteria to integrin α2/β1 was necessary for the tumor-promoting activity of S. moorei. In addition, we have preliminarily explored the ligand on the surface of S. moorei that binds to integrin α2/β1 on CRC cells and speculated that S. moorei cellwall protein Cna B-type domain-containing protein might bind to integrin α2/β1. According to the different binding characteristics of integrins, they can be divided into four types: leukocyte adhesion integrin, RGD binding integrin, collagen binding integrin, and laminin binding integrin. Integrin α2/β1 is one of collagen binding integrins which expressed on epithelial cells[45]. Through the docking model (Figure 7D), we have found Cna B-type domain-containing protein conjugate with I domain (AA:140-359) of integrinα2β1 which is also the key domain in binding of collagen[46, 47]. Not only that, we further discovered Cna B-type domain-containing protein conjugated to integrin β1 and promoted CRC cells growth which determined its role in cancer progression (Figure 7E-F). In our research, we found RGD peptides, which had been reported regulating conjugation of collagen and integrin[48], impended integrin downstream signal provoked by S. moorei. Thus, we speculated Cna B-type domain-containing protein might play roles like collagen which activated cancer cell proliferation and migration.
Conclusions
In summary, we found that S. moorei was enriched in fecal samples from CRC patients compared to healthy subjects and was also enriched in CRC tissues from ApcMin/+ mice compared to matched non-tumor tissues. S. moorei abundance was significantly correlated with tumor staging. S. moorei cellwall protein Cna B-type domain-containing protein interacted with integrin α2/β1, which was overexpressed on CRC cells, leading to the activation of PI3K-AKT-mTOR-C-myc pathway via phospho-FAK; thus, promoting cell growth and tumor progression. This work explored the effect of S. moorei on the progression of CRC and identified a potential molecular mechanism by which S. moorei promoted CRC progression. This study suggests that S. moorei might be a new potential target for CRC prevention, diagnosis, and treatment.
Supplementary Material
Supplementary figures and tables 1-4, 11.
Supplementary table 5.
Supplementary table 6.
Supplementary table 7.
Supplementary table 8.
Supplementary table 9.
Supplementary table 10.
Acknowledgements
We are deeply appreciative of the participants who donated specimens and the colleagues in our laboratory for their invaluable assistance and technical support.
Grant support
This work was supported by Shenzhen Science and Technology Innovation Commission (No.2021293 and No.202231) and Open Fund of State Key Laboratory of Chemical Oncogenomics (No. SKLCO202203).
Ethics approval and consent to participate
Involving human participants and animal experiments in this study was approved by the Bioethics committee of Tsinghua Shenzhen International Graduate School. All subjects gave written informed consent in accordance with the Declaration of Helsinki before their inclusion in the study and extensive efforts were made to ensure minimal suffering of the included animals.
Consent for publication
All authors have approved the submission of the manuscript for publication and agree to be accountable for all aspects of the manuscript.
Data sharing statement
The data that support the findings of this study are available on request from the corresponding author.
Author contributions
YC is the principal investigator. YC, HY, and YYJ designed and supervised the research. YC, YJZ and YYZ conducted the majority of the molecular and cellular experiments. YQ collected the clinical specimens. YC, CQ and GY performed the animal experiments. TTF and YYZ conducted data management and visualization. QSS conducted statistical analysis. YC was a major contributor in writing the manuscript. HY and YYJ revised the manuscript critically. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wong CC, Yu J. Gut microbiota in colorectal cancer development and therapy. Nat Rev Clin Oncol. 2023;20:429-52
2. Qu R, Zhang Y, Ma Y, Zhou X, Sun L, Jiang C. et al. Role of the Gut Microbiota and Its Metabolites in Tumorigenesis or Development of Colorectal Cancer. Adv Sci (Weinh). 2023;10:e2205563
3. Ralser A, Dietl A, Jarosch S, Engelsberger V, Wanisch A, Janssen KP. et al. Helicobacter pylori promotes colorectal carcinogenesis by deregulating intestinal immunity and inducing a mucus-degrading microbiota signature. Gut. 2023;72:1258-70
4. Hou H, Chen D, Zhang K, Zhang W, Liu T, Wang S. et al. Gut microbiota-derived short-chain fatty acids and colorectal cancer: Ready for clinical translation? Cancer Lett. 2022;526:225-35
5. Jiang SS, Xie YL, Xiao XY, Kang ZR, Lin XL, Zhang L. et al. Fusobacterium nucleatum-derived succinic acid induces tumor resistance to immunotherapy in colorectal cancer. Cell Host Microbe. 2023;31:781-97.e9
6. Lu Y, Li Z, Peng X. Regulatory effects of oral microbe on intestinal microbiota and the illness. Front Cell Infect Microbiol. 2023;13:1093967
7. Schmidt TS, Hayward MR, Coelho LP, Li SS, Costea PI, Voigt AY. et al. Extensive transmission of microbes along the gastrointestinal tract. Elife. 2019;8:e42693
8. Mo S, Ru H, Huang M, Cheng L, Mo X, Yan L. Oral-Intestinal Microbiota in Colorectal Cancer: Inflammation and Immunosuppression. J Inflamm Res. 2022;15:747-59
9. Flemer B, Warren RD, Barrett MP, Cisek K, Das A, Jeffery IB. et al. The oral microbiota in colorectal cancer is distinctive and predictive. Gut. 2018;67:1454-63
10. Bergsten E, Mestivier D, Donnadieu F, Pedron T, Barau C, Meda LT. et al. Parvimonas micra, an oral pathobiont associated with colorectal cancer, epigenetically reprograms human colonocytes. Gut Microbes. 2023;15:2265138
11. Wang X, Jia Y, Wen L, Mu W, Wu X, Liu T. et al. Porphyromonas gingivalis Promotes Colorectal Carcinoma by Activating the Hematopoietic NLRP3 Inflammasome. Cancer Res. 2021;81:2745-59
12. Long X, Wong CC, Tong L, Chu ESH, Ho Szeto C, Go MYY. et al. Peptostreptococcus anaerobius promotes colorectal carcinogenesis and modulates tumour immunity. Nat Microbiol. 2019;4:2319-30
13. Barrak I, Stájer A, Gajdács M, Urbán E. Small, but smelly: the importance of Solobacterium moorei in halitosis and other human infections. Heliyon. 2020;6:e05371
14. Haraszthy VI, Zambon JJ, Sreenivasan PK, Zambon MM, Gerber D, Rego R. et al. Identification of oral bacterial species associated with halitosis. J Am Dent Assoc. 2007;138:1113-20
15. Liu WJ, Xiao M, Yi J, Li Y, Kudinha T, Xu YC. First case report of bacteremia caused by Solobacterium moorei in China, and literature review. BMC Infect Dis. 2019;19:730
16. Sárvári KP, Sántha D, Kovács R, Körmöndi S, Pető Z, Vereb T. et al. Six cases of Solobacterium moorei isolated alone or in mixed culture in Hungary and comparison with previously published cases. Anaerobe. 2020;65:102241
17. George N, Flamiatos E, Kawasaki K, Kim N, Carriere C, Phan B. et al. Oral microbiota species in acute apical endodontic abscesses. J Oral Microbiol. 2016;8:30989
18. Bachtiar BM, Soeroso Y, Sunarto H, Maitimu FC, Bachtiar EW. Relationships between Solobacterium moorei and Prevotella intermedia in subgingival microbiota of periodontitis patients with halitosis: A preliminary study using qPCR. Saudi Dent J. 2022;34:211-9
19. Vancauwenberghe F, Dadamio J, Laleman I, Van Tornout M, Teughels W, Coucke W. et al. The role of Solobacterium moorei in oral malodour. J Breath Res. 2013;7:046006
20. LeBel G, Haas B, Adam AA, Veilleux MP, Lagha AB, Grenier D. Effect of cinnamon (Cinnamomum verum) bark essential oil on the halitosis-associated bacterium Solobacterium moorei and in vitro cytotoxicity. Arch Oral Biol. 2017;83:97-104
21. Yang K, Wang Y, Zhang S, Zhang D, Hu L, Zhao T. et al. Oral Microbiota Analysis of Tissue Pairs and Saliva Samples From Patients With Oral Squamous Cell Carcinoma - A Pilot Study. Front Microbiol. 2021;12:719601
22. Yu J, Feng Q, Wong SH, Zhang D, Liang QY, Qin Y. et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut. 2017;66:70-8
23. Yachida S, Mizutani S, Shiroma H, Shiba S, Nakajima T, Sakamoto T. et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat Med. 2019;25:968-76
24. Avuthu N, Guda C. Meta-Analysis of Altered Gut Microbiota Reveals Microbial and Metabolic Biomarkers for Colorectal Cancer. Microbiol Spectr. 2022;10:e0001322
25. Yu S, Wang X, Li Z, Jin D, Yu M, Li J. et al. Solobacterium moorei promotes the progression of adenomatous polyps by causing inflammation and disrupting the intestinal barrier. J Transl Med. 2024;22:169
26. Wu S, Sun C, Li Y, Wang T, Jia L, Lai S. et al. GMrepo: a database of curated and consistently annotated human gut metagenomes. Nucleic Acids Res. 2020;48:D545-d53
27. Nani BD, Lima PO, Marcondes FK, Groppo FC, Rolim GS, Moraes AB. et al. Changes in salivary microbiota increase volatile sulfur compounds production in healthy male subjects with academic-related chronic stress. PLoS One. 2017;12:e0173686
28. Afriyie-Asante A, Dabla A, Dagenais A, Berton S, Smyth R, Sun J. Mycobacterium tuberculosis Exploits Focal Adhesion Kinase to Induce Necrotic Cell Death and Inhibit Reactive Oxygen Species Production. Front Immunol. 2021;12:742370
29. Wang YL, Zhu BJ, Qi ZZ, Wang HJ, Zhou XD. Akt1 enhances CA916798 expression through mTOR pathway. PLoS One. 2013;8:e62327
30. Schrader A, Bentink S, Spang R, Lenze D, Hummel M, Kuo M. et al. High Myc activity is an independent negative prognostic factor for diffuse large B cell lymphomas. Int J Cancer. 2012;131:E348-61
31. Liu Y, Song Z, Liu Y, Ma X, Wang W, Ke Y. et al. Identification of ferroptosis as a novel mechanism for antitumor activity of natural product derivative a2 in gastric cancer. Acta Pharm Sin B. 2021;11:1513-25
32. Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10:1523
33. Li M, Wang Y, Li M, Wu X, Setrerrahmane S, Xu H. Integrins as attractive targets for cancer therapeutics. Acta Pharm Sin B. 2021;11:2726-37
34. Luo J, Yao JF, Deng XF, Zheng XD, Jia M, Wang YQ. et al. 14, 15-EET induces breast cancer cell EMT and cisplatin resistance by up-regulating integrin αvβ3 and activating FAK/PI3K/AKT signaling. J Exp Clin Cancer Res. 2018;37:23
35. Pandey H, Tang DWT, Wong SH, Lal D. Gut Microbiota in Colorectal Cancer: Biological Role and Therapeutic Opportunities. Cancers (Basel). 2023;15:866
36. Scheible M, Nguyen CT, Luong TT, Lee JH, Chen YW, Chang C. et al. The Fused Methionine Sulfoxide Reductase MsrAB Promotes Oxidative Stress Defense and Bacterial Virulence in Fusobacterium nucleatum. mBio. 2022;13:e0302221
37. Mu W, Jia Y, Chen X, Li H, Wang Z, Cheng B. Intracellular Porphyromonas gingivalis Promotes the Proliferation of Colorectal Cancer Cells via the MAPK/ERK Signaling Pathway. Front Cell Infect Microbiol. 2020;10:584798
38. Sun L, Zhang Y, Cai J, Rimal B, Rocha ER, Coleman JP. et al. Bile salt hydrolase in non-enterotoxigenic Bacteroides potentiates colorectal cancer. Nat Commun. 2023;14:755
39. Cooper J, Giancotti FG. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell. 2019;35:347-67
40. Zhou J, Yi Q, Tang L. The roles of nuclear focal adhesion kinase (FAK) on Cancer: a focused review. J Exp Clin Cancer Res. 2019;38:250
41. Wu X, Wang J, Liang Q, Tong R, Huang J, Yang X. et al. Recent progress on FAK inhibitors with dual targeting capabilities for cancer treatment. Biomed Pharmacother. 2022;151:113116
42. Zhang J, Li X, Zhou Y, Lin M, Zhang Q, Wang Y. FNBP1 Facilitates Cervical Cancer Cell Survival by the Constitutive Activation of FAK/PI3K/AKT/mTOR Signaling. Cells. 2023;12:1964
43. Wang R, Wang S, Li Z, Luo Y, Zhao Y, Han Q. et al. PLEKHH2 binds β-arrestin1 through its FERM domain, activates FAK/PI3K/AKT phosphorylation, and promotes the malignant phenotype of non-small cell lung cancer. Cell Death Dis. 2022;13:858
44. Ke S, Liu Z, Wang Q, Zhai G, Shao H, Yu X. et al. FAM107A Inactivation Associated with Promoter Methylation Affects Prostate Cancer Progression through the FAK/PI3K/AKT Pathway. Cancers (Basel). 2022;14:3915
45. Pang X, He X, Qiu Z, Zhang H, Xie R, Liu Z. et al. Targeting integrin pathways: mechanisms and advances in therapy. Signal Transduct Target Ther. 2023;8:1
46. Kamata T, Puzon W, Takada Y. Identification of putative ligand binding sites within I domain of integrin alpha 2 beta 1 (VLA-2, CD49b/CD29). J Biol Chem. 1994;269:9659-63
47. Tuckwell D, Calderwood DA, Green LJ, Humphries MJ. Integrin alpha 2 I-domain is a binding site for collagens. J Cell Sci. 1995;108( Pt 4):1629-37
48. Casal JI, Bartolomé RA. RGD cadherins and α2β1 integrin in cancer metastasis: A dangerous liaison. Biochim Biophys Acta Rev Cancer. 2018;1869:321-32
Author contact
![]() Corresponding authors: Dr. Yan Chen, E-mail: chenyanedu.cn; Prof. Yue Hao, E-mail: yuehaoedu.cn; Prof. Yuyang Jiang, E-mail: jiangyytsinghua.edu.cn.
Corresponding authors: Dr. Yan Chen, E-mail: chenyanedu.cn; Prof. Yue Hao, E-mail: yuehaoedu.cn; Prof. Yuyang Jiang, E-mail: jiangyytsinghua.edu.cn.