Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Result
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(7):3304-3321. doi:10.7150/ijbs.126649 This issue Cite
Research Paper
Macrophage NEDD4L restrains liver fibrosis by preventing scar-associated macrophage expansion via ubiquitination of phospho-SMAD3
Yanghuan He1,5#, Shujun Ge2#, Shijia Ling2#, Siting Yang2#, Feiran Yang2, Xinyi Liu2, Siyue Dong2, Yingfen Chen3,4, Ziling Zhang2, Yue Zhou3,4, Seonghwan Hwang6, Seung-Jin Kim7, Peng Wang8 ![]() , Yong He2,3,4
, Yong He2,3,4 ![]() , Yuanwen Chen1,5
, Yuanwen Chen1,5 ![]()
1. Department of Gastroenterology, Huadong Hospital, Fudan University, Shanghai, China.
2. School of Chinese Materia Medica, Nanjing University of Chinese Medicine, Nanjing, China.
3. State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica (SIMM), Chinese Academy of Sciences, Shanghai, China.
4. University of Chinese Academy of Sciences, Beijing, China.
5. Shanghai Key Laboratory of Clinical Geriatric Medicine, Shanghai Institute of Geriatrics and Gerontology, Shanghai, China.
6. College of Pharmacy and Research Institute for Drug Development, Pusan National University, Busan, Republic of Korea.
7. Department of Biochemistry, College of Natural Sciences, Kangwon National University, Chuncheon, Republic of Korea.
8. Department of Hepatobiliary Medicine, Shanghai Eastern Hepatobiliary Surgery Hospital, Naval Medical University, Shanghai, China.
# Yanghuan He, Shujun Ge, Shijia Ling and Siting Yang contributed equally to this work.
Received 2025-10-13; Accepted 2026-2-18; Published 2026-3-17
Abstract

Background: Liver fibrosis is characterized by excessive extracellular matrix deposition and hepatic stellate cell (HSC) activation, driven by chronic liver injury and inflammation. Macrophages play dual roles in fibrogenesis; the dynamic balance between pro-fibrotic and anti-fibrotic subsets is critical in determining the progression or regression of the disease. NEDD4L, an E3 ubiquitin ligase, is well-known to be involved in cell biological processes by promoting protein degradation, yet its role in macrophages and liver fibrosis remains poorly understood.
Methods: Myeloid cell-specific Nedd4l knockout (Nedd4lf/f Lyz-Cre+, Nedd4lΔMye) were generated, and subjected to carbon tetrachloride (CCl4) and choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD)-induced experimental liver fibrosis models.
Results: Single-cell RNA sequencing and transcriptomic analyses revealed significant upregulation of NEDD4L in macrophages from human and murine fibrotic livers. Strikingly, myeloid cell-specific Nedd4l deficiency exacerbated liver fibrosis in both mouse models, as evidenced by increased collagen deposition and elevated expression of fibrogenic genes in Nedd4lΔMye mice. Notably, mice with Nedd4l deficient in macrophages had more pro-fibrotic scar-associated macrophage (SAM) infiltration compared with Nedd4lf/f mice in two experimental models. More interestingly, coculture in vitro experiments further verified that TGF-β1-treated Nedd4l deficient macrophages promoted HSC activation due to greater activation of SMAD3 signaling. Mechanistically, NEDD4L directly ubiquitinated phosphorylated SMAD3 and led to its degradation, thus limiting TGF-β1/SMAD3 signaling in macrophages. Moreover, hepatic levels of NEDD4L were significantly elevated in patients with liver fibrosis, positively correlating with hepatic levels of several fibrogenic genes.
Conclusions: NEDD4L serves as a critical negative regulator of liver fibrosis by restraining profibrotic SAM expansion through ubiquitination and degradation of p-SMAD3 in macrophages. These findings highlight that targeting the ubiquitin-proteasome system as a potential therapeutic strategy for the treatment of fibrotic disease.
Keywords: macrophage differentiation, ubiquitination, liver fibrogenesis, p-SMAD3.
Introduction
Liver fibrosis, a pathological consequence of chronic liver injury caused by, e.g., viral hepatitis, alcohol abuse, or metabolic dysfunction, is characterized by excessive extracellular matrix (ECM) deposition and activation of hepatic stellate cells (HSCs) [1]. Despite advances in understanding its pathogenesis, effective anti-fibrotic therapies remain limited [2, 3]. Earlier studies posited that the central role of macrophages in orchestrating fibrogenesis through their dynamic polarization states—pro-inflammatory M1 macrophages driving tissue injury, while anti-inflammatory M2 macrophages promoting fibrosis. Mounting evidence demonstrate heterogeneous differentiation of hepatic macrophages in liver fibrosis, with distinct subsets exhibiting specialized functions [4, 5]. Recent single-cell RNA sequencing (scRNA-seq) studies have further refined this classification, identifying novel subpopulations such as scar-associated macrophages (SAMs) and TREM2+ macrophages including lipid-associated macrophages (LAMs) that play decisive roles in disease progression [6, 7]. However, the molecular mechanisms regulating macrophage plasticity during liver fibrogenesis are incompletely understood.
NEDD4L (neural precursor cell expressed, developmentally downregulated 4-like), a member of the NEDD4 family of E3 ubiquitin ligases, plays a vital role in regulating protein degradation and fundamental cellular processes [8]. The NEDD4L protein comprises a C2 domain, three to four WW domains, and a HECT domain [9]. These domains collectively mediate substrate binding and catalyze ubiquitin conjugation to target proteins, triggering their degradation via the proteasome pathway. Most identified targets of NEDD4L are membrane proteins, including ion channels and transporters [10]. Recently, accumulating studies suggest that NEDD4L in hepatocytes plays an important role in the pathogenesis of metabolic dysfunction-associated steatotic liver disease (MASLD). For example, NEDD4L in hepatocytes leads to the degradation of lysosomal-associated protein transmembrane 5 (LAPTM5), which interacts with CDC42 and promotes its degradation through a lysosome-dependent manner under the stimulation of palmitic acid, finally exacerbating MASLD by activating mitogen-activated protein kinase signaling pathway [11]. In addition, NEDD4L targets thioredoxin interacting protein (TXNIP) in hepatocytes, which elevates CHOP, a major regulator of ER stress-mediated apoptosis, thereby affecting the development of MASLD [12]. Another interesting study found that hepatic Fas apoptotic inhibitory molecule 2 (FAIM2) is degraded by NEDD4L through the catalysis of K48-linked ubiquitination, leading to its protein downregulation. Such downregulation aggravates MASLD by promoting the autophagic degradation of CREB-regulated transcription coactivator 2 (CRTC2), a prominent regulator of lipid metabolism [13]. Although these studies highlight the key role of NEDD4L in hepatocytes during MASLD progression, the functional role of NEDD4L in macrophages remains obscure.
Cytokines of the transforming growth factor-β (TGF-β) family signal through serine/threonine kinase receptors to control cellular behavior and fate [14]. These signals are propagated via the transcription factors Smad2/3 downstream of TGF-β1, activin, and nodal receptors, and Smad1/5/8 downstream of bone morphogenetic protein (BMP) receptors [15]. The TGF-β family of multifunctional cytokines plays a pivotal role in the pathogenesis of metabolic dysfunction-associated steatohepatitis (MASH), especially liver fibrosis development [16]. More interestingly, TGF-β signaling orchestrates multiple facets of macrophage biology, including differentiation, polarization, inflammatory responses, tissue homeostasis, and tumorigenesis [17]. For example, genetic disruption of TGF-β signaling compromises the development of alveolar and intestinal macrophages [18, 19]. Macrophage-derived TGF-β signaling drives immunosuppression in the tumor microenvironment, facilitating immune evasion across multiple cancer types [20]. Importantly, emerging evidence indicates that NEDD4L modulates the TGF-β signaling pathway by ubiquitinating key downstream substrates [21, 22]. The roles of macrophage NEDD4L have been investigated in diverse pathological contexts. For instance, in viral infections, NEDD4L has been shown to promote antiviral innate immunity by catalyzing K29-linked cysteine ubiquitination of TRAF3, thereby enhancing type I interferon production [23]. Conversely, in intracerebral hemorrhage (ICH)-induced brain injury, NEDD4L exacerbates neuroinflammation and brain damage by facilitating M1 macrophage polarization via the TRAF3/TBK1 signaling pathway [24]. In this current study, by generating genetic knockout mouse models and ubiquinone profiling, we demonstrate that macrophage NEDD4L serves as a critical negative regulator of liver fibrosis by restraining profibrotic SAM-related macrophage expansion through ubiquitination and degradation of p-SMAD3 in macrophages. Our findings unveil novel therapeutic targets for the treatment of chronic inflammation-driven liver fibrosis by harnessing the ubiquitin-proteasome system in immune modulation.
Materials and Methods
Human liver samples
The paraffin-embedded human liver samples were collected from Shanghai Eastern Hepatobiliary Surgery Hospital, Naval Medical University. The study protocol received ethical approval from the hospital's Institutional Review Board (Approval No. EHBHKY2020-K-048). The baseline demographics and clinical characteristics of the patient cohorts are summarized in Supplementary Table S1.
Animal models and dietary interventions
Myeloid cell-specific Nedd4l Knockout (Nedd4lΔMye) mice were generated by crossing Nedd4l flox/flox mice with Lyz-Cre transgenic mice. Littermate control mice (Nedd4lf/f) were served as controls. For the carbon tetrachloride (CCl4)-induced liver fibrosis model, mice were intraperitoneally injected with 2 ml/kg weight of 10% CCl4 (MACKLIN, catalog O815211, Shanghai, China) in olive oil (MACKLIN, catalog C805325, Shanghai, China) twice per week for 4 weeks. For MASH-related liver fibrosis model, mice were fed a choline-deficient L-amino acid-defined high-fat diet (CDAHFD, A06071302; Research Diets) for 2 weeks. All animal studies were conducted under protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the Shanghai Institute of Materia Medica, Chinese Academy of Sciences and the Institutional Animal Care and Use Committee of Fudan University (IACUC-2023-012). Mice were housed under a 12-hours light/dark cycle with ad libitum access to food and water.
Differentiation of primary bone marrow-derived macrophages (BMDMs)
Irrigate the bone marrow cavity of the mouse femur and tibia with sterile PBS using a syringe, then grind the tissue and filter it through a 70-μm cell strainer. Next, the samples were centrifuged at 1600 rpm, 4 °C, for 5 minutes to precipitate the bone marrow cells (BM). The red blood cells were lysed using red blood cell lysis buffer, resuspended cells in PBS, and filtered through a 70-μm cell strainer. BM cells were seeded into 24-well cell culture plates and added conditioned medium containing 20 ng/ml MCSF (1640 medium, 10% FBS + 1% PS), and cultured in a 37 °C, 5% CO₂ incubator. On day 3, add the same fresh conditioned medium and continue culturing. After day 7, the cells were washed with PBS and used for subsequent experiments.
Plasmid construction
The pCDH-CMV-HIS-UB-MCS-3Flag-EF1-CopGFP-T2A-Puro plasmid was generated by inserting the UB into the XbaI/AsisI sites of the multicoloning site (MCS) of the pCDH vector backbone. The plasmid was commercially synthesized and subcloned by YiXueSheng Biosciences Inc (Shanghai, China). The final construct was validated through double restriction enzyme digestion analysis and full-length sequencing using primers specific to the CMV promoter.
The pcDNA3.1-SMAD3-Myc plasmid was generated by inserting the SMAD3 into the BamHI/EcoRI and pcDNA3.1sites of the multicoloning site (MCS) of the pCDH vector backbone. The plasmid was commercially synthesized and subcloned by YiXueSheng Biosciences Inc (Shanghai, China). The final construct was validated through double restriction enzyme digestion analysis and full-length sequencing using primers specific to the CMV promoter and the bGh terminator.
The pcDNA3.1-Nedd4l-Flag plasmid was generated by inserting the NEDD4L into the BamHI/EcoRI and pcDNA3.1sites of the multicoloning site (MCS) of the pCDH vector backbone. The plasmid was commercially synthesized and subcloned by WeiZhenSheng Biosciences Inc (Shanghai, China). The final construct was validated through double restriction enzyme digestion analysis and full-length sequencing using primers specific to the CMV promoter and the bGh terminator.
In vitro co-culture assays
BMDMs were differentiated as described above and then stimulated with or without 10 ng/mL TGF-β1 for 24 hours. The culture supernatant was then removed, and the cells were washed and incubated in serum-free DMEM for another 12 hours to generate conditioned medium (CM). The CM was then collected, centrifuged to remove cellular debris, and mixed at a 1:1 ratio with fresh RPMI-1640 medium. This mixture was applied to pre-plated, unactivated LX-2 hepatic stellate cells (HSCs). Co-cultures were maintained for various durations, as indicated, after which cells and/or supernatants were harvested for subsequent analysis.
Biochemical assays
Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were determined using ALT Detection Kit (Nanjing Jiancheng Bioengeering Institute, China, catalog C009-3-1) and AST Detection Kit (Nanjing Jiancheng Bioengeering Institute, China, catalog C010-3-1) according to the manufacturer's instruction.
Immunohistochemistry and Sirius red staining
For Sirius red staining, the paraffin-embedded sections were dewaxed in xylene and stained with PICRO-RED STAINING SOLUTION (Phygene, catalog 20230310, Fuzhou, China) for 1 hour. For immunohistochemistry, sections were subjected to antigen retrieval with citrate buffer (Invitrogen, catalog 005000, CA, USA). After incubating at 3% H2O2, sections were blocked with 3% normal goat serum buffer (NGS). Then, the sections were incubated with primary antibodies overnight at 4 °C and incubated with secondary antibodies (Cell Signaling Technology, Danvers, MA, catalog 8814S or 8125S) at room temperature for 1 hour. ImmPACT DAB Substrate Kit (Vector Laboratories, catalog ZK1018, CA, USA) or ImmPACT Red Substrate Kit (Vector Laboratories, catalog ZJ1205, CA, USA) were used to visualize the staining. Primary antibodies used were listed below: F4/80 (Cell Signaling Technology catalog 70076S, Danvers, MA, USA), Myeloperoxidase (Biocare medical, catalog APR023AA, CA, USA), CD3 (Cell Signaling Technology catalog 78588S, Danvers, MA, USA), α-SMA (Cell Signaling Technology, catalog 19245S, Danvers, MA, USA), Col1α1 (Cell Signaling Technology, catalog 72026S, CST, Danvers, MA), NEDD4L (Cell Signaling Technology, catalog 4013, Danvers, MA, USA), p-Smad3 (ABclonal Technology, catalog AP1263, Wuhan, China), FABP5 (ABclonal Technology, catalog A27255, Wuhan, China), CD9 (ABclonal Technology, catalog A19027, Wuhan, China), CD163 (ABclonal Technology, catalog A8383, Wuhan, China), CD206 (ABclonal Technology, catalog A21014, Wuhan, China), TLR1 (ABclonal Technology, catalog A0997, Wuhan, China), Trem2 (Abcam, catalog AB305103 Cambridge, UK). Images were obtained using the Olympus BX43 microscope.
Immunofluorescence
Fresh liver tissues were frozen and embedded for sectioning. After overnight fixation with 4% PFA at 4 °C, 3% NGS was used to block for one hour at room temperature. Next, primary antibodies were incubated overnight at 4°C, and secondary antibodies (Alexa Fluor 488 or 555 goat anti-rabbit IgG [H+L], Cell Signaling Technology, catalog 4412S or 4413S; Alexa Fluor 488 or 555 goat anti-mouse IgG [H+L], Cell Signaling Technology, catalog 4408S or 4409S, Danvers, MA, USA) were incubated for 1 hour at room temperature. TrueVIEW Autofluorescence Quenching Kit (Vector Laboratories, catalog ZK0818, CA, USA) was used to eliminate non-specific fluorescence. Nuclear staining was performed using 4', 6'-diamino-2-phenylindole (DAPI) (Beyotime, catalog P0131, Shanghai, China). Images were captured with Olympus APEXVIEW APX100 microscope. The following primary antibodies were used: NEDD4L (Cell Signaling Technology, catalog 4013, Danvers, MA, USA), IBA1 (catalog No. MABN92, Merck KGaA, Darmstadt, Germany), CD206 (ABclonal Technology, catalog A21014, Wuhan, China), FABP5 (ABclonal Technology, catalog A27255, Wuhan, China).
Total RNA isolation and reverse transcription quantitative PCR (RT-qPCR)
Total RNA was extracted using RNA isolater Total RNA Extraction Reagent (Vazyme, catalog 7E0131K3, Nanjing, China), and single-stranded cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, catalog 2816898, Waltham, MA). Gene expression was determined by qPCR using ChamQ Universal SYBR qPCR Master Mix (Vazyme, catalog 7E751K3, Nanjing, China) and a QuantStudio 5 Instrument (Thermo Fisher Scientific, Waltham, MA). The levels of the rRNA 18s were used as an internal control. Each test was performed in triple replication and the 2-∆∆Ct method was used to calculate the expression of mRNA. The primers used for RT-qPCR are listed in Supplementary Table 2.
Co-immunoprecipitation (Co-IP) assay
Co-immunoprecipitation assays were performed to investigate the interaction between [Protein A] and [Protein B]. Briefly, HEK293T cells transfected with relevant plasmids were lysed in IP lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 10% glycerol) supplemented with a protease inhibitor cocktail (Thermo Fisher Scientific, catalog 88804, Waltham, MA, USA) on ice for 5 minutes. The lysates were centrifuged for 10 minutes at 4 °C to remove cellular debris. The supernatant was then incubated with 2 μg of Mouse anti DDDDK-Tag (ABclonal Technology, catalog AE005, Wuhan, China) or Myc-Tag Rabbit mAb (ABclonal Technology, catalog AE070, Wuhan, China) overnight at 4 °C with gentle rotation. Subsequently, 25 μL of Protein A/G Magnetic Beads (Thermo Fisher Scientific, catalog 88804, Waltham, MA, USA) were added and incubated for an additional 1 hour at 4 °C. The beads were then washed five times with ice-cold lysis buffer. Finally, the bound proteins were eluted by boiling in 2× Laemmli SDS sample buffer at 95 °C for 10 minutes. The collected protein samples were used for subsequent experiments.
Western blotting
Proteins were extracted from liver tissues or cells using RIPA Buffer (Thermo Fisher Scientific, catalog YH374135, Waltham, MA, USA) containing Halt Protease and Phosphatase Inhibitors (Thermo Fisher Scientific, catalog 78447, Waltham, MA, USA). The samples were loaded into polyacrylamide gels (Absin, catalog 220A019, Shanghai, China) and then transferred onto nitrocellulose membranes (Merck, catalog 0000208128, Darmstadt, Germany). The nitrocellulose membranes were blocked with 1% BSA and incubated with antibodies overnight at 4 °C. After incubation with anti-rabbit or anti-mouse IgG HRP-linked second antibody (Cell Signaling Technology, catalog 7074S or 7076S, Danvers, MA, USA), protein bands were visualized with SuperSignal Maximum Sensitivity Substrate (Thermo Fisher Scientific, catalog WG328673, Waltham, MA, USA). The following antibodies were used: Col1α1 (Abcam, catalog AB260043, Cambridge, UK,), α-SMA (Thermo Fisher Scientific, catalog 14-9760-82, Waltham, MA, USA), NEDD4L (Cell Signaling Technology, catalog 4013, Danvers, MA, USA), p38 (Cell Signaling Technology, catalog 8690S, Danvers, MA, USA), p-P38 (Cell Signaling Technology, catalog 4511, Danvers, MA, USA), ERK (Cell Signaling Technology, catalog 4695S, Danvers, MA, USA), p-ERK (Cell Signaling Technology, catalog 4370S, Danvers, MA, USA), p-Smad2/3 (Cell Signaling Technology, catalog 8828S, Danvers, MA, USA), Smad2/3 (Cell Signaling Technology, catalog 8685S, Danvers, MA, USA), p-Smad3 (ABclonal Technology, catalog AP1263, Wuhan, China), HRP-conjugated Mouse anti-Rabbit IgG Light Chain (ABclonal Technology, catalog AS061, Wuhan, China), Mouse anti DDDDK-Tag (ABclonal Technology, catalog AE005, Wuhan, China), HA-Tag Rabbit mAb (ABclonal Technology, catalog AE105, Wuhan, China), Myc-Tag Rabbit mAb (ABclonal Technology, catalog AE070, Wuhan, China).
Statistical analysis
Data are expressed as the mean ± SEM and were analyzed using GraphPad Prism software (version 9.4.1; GraphPad Software). To compare values obtained from two groups, the Student t test was performed; values from multiple groups were compared using one-way ANOVA. p<0.05 was considered significant.
Result
Macrophage NEDD4L is elevated during liver fibrosis development
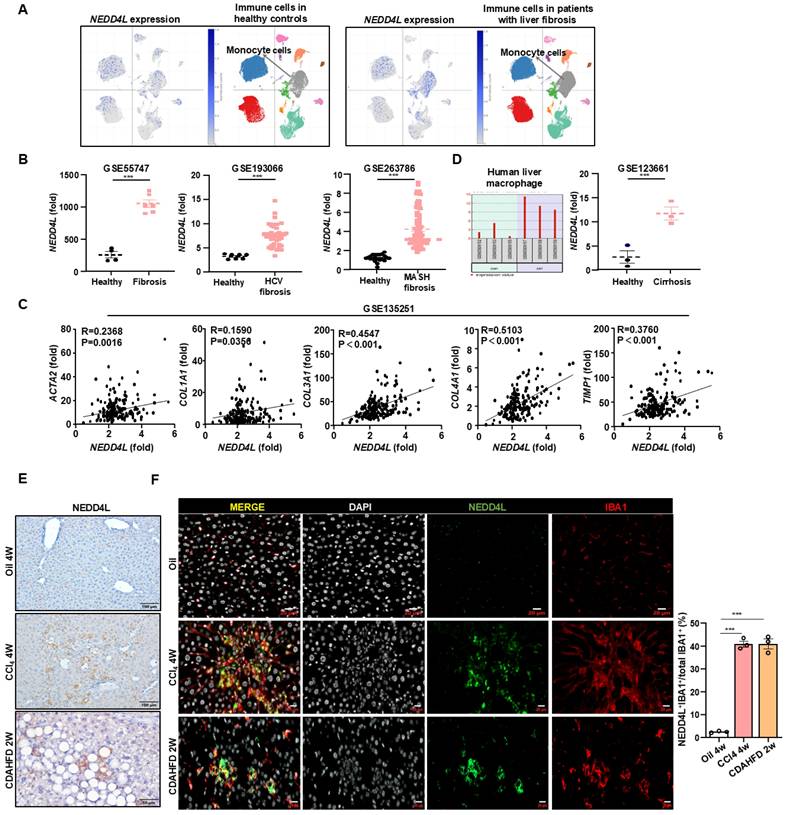
To understand the role of NEDD4L in liver fibrosis, we first analyzed public single-cell RNA sequencing (scRNA-seq) database (https://singlecell.broadinstitute.org). Initial scRNA-seq analysis of healthy human livers revealed only minimal Nedd4l expression across immune cell populations. In contrast, analysis of samples from patients with liver fibrosis showed a marked induction of NEDD4L. Such upregulation was observed in several immune populations, and was most pronounced within monocyte-derived macrophages (Fig. 1A, Supporting Fig. S1A). This striking, cell-type-associated induction indicates a disease-specific transcriptional reprogramming, likely reflecting the functional adaptation of this myeloid subset during liver fibrogenesis. To assess the generality of this response, we further performed a cross-database analysis of transcriptomic datasets from fibrotic livers of diverse etiologies. Consistent upregulation of NEDD4L was observed in HCV-induced fibrosis (GSE193066) and metabolic dysfunction-associated steatotic liver disease (MASLD)-driven fibrosis (GSE263786) compared to healthy controls (Fig. 1B). In the MASLD fibrosis dataset (GSE135251), NEDD4L expression showed a positive correlation with several core pro-fibrotic genes including ACTA2, COL1A1, COL3A1, COL4A1, and TIMP1, suggesting a potential relationship between NEDD4L and liver fibrogenesis (Fig. 1C). Moreover, analysis of isolated hepatic macrophages from cirrhotic patients confirmed elevated NEDD4L expression at the cellular levels, reinforcing its link to advanced disease (Fig. 1D). This induction was conserved across species, as elevated NEDD4L was also observed in murine models of carbon tetrachloride (CCl4)-induced and choline-deficient, L-amino acid-defined high-fat diet (CDAHFD)-induced liver fibrosis (Fig. 1E). In fibrotic livers induced by both CCl4 and CDAHFD, immunofluorescence co-staining revealed a specific population of IBA1⁺ macrophages within the fibrotic lesions. Critically, NEDD4L expression was predominantly co-localized with these cells, which collectively established the myeloid cell-specific upregulation of NEDD4L during liver fibrosis (Fig. 1F). Taken together, these multi-level analyses—spanning single-cell RNA sequencing, cross-dataset validation, molecular correlation, and protein-level visualization—consistently demonstrate that NEDD4L expression is significantly upregulated in hepatic macrophages in both human and experimental mouse liver fibrosis, highlighting its potential role as a key regulator in the fibrotic microenvironment.
Macrophage NEDD4L is upregulated in human and murine liver fibrosis. (A) scRNA-seq analysis showing NEDD4L expression in immune cells from healthy and fibrotic human livers. (B) Cross-dataset validation of NEDD4L upregulation in HCV- and MASLD-induced fibrosis (GSE55747, GSE193066, and GSE263786). (C) The correlations between hepatic NEDD4L levels and liver fibrogenic genes in patients with liver fibrosis were analyzed from the GSE135251 dataset. (D) NEDD4L expression in isolated hepatic macrophages from cirrhosis patients (GSE123661). (E) Representative images of NEDD4L staining (Scale bar: 200 μm) in the livers of control and liver fibrosis mouse models are shown. (F) Representative immunofluorescence staining of IBA1 (red), NEDD4L (green), nuclei (white) (Scale bar: 20 μm) in the livers of CCl4 and CDAHFD-induced liver fibrosis mice are shown. The percentage of positive cells was quantified. Values represent mean ± SEM. ***p<0.001.
Myeloid cell-specific knockout of Nedd4l exacerbates CCl4-induced liver fibrosis
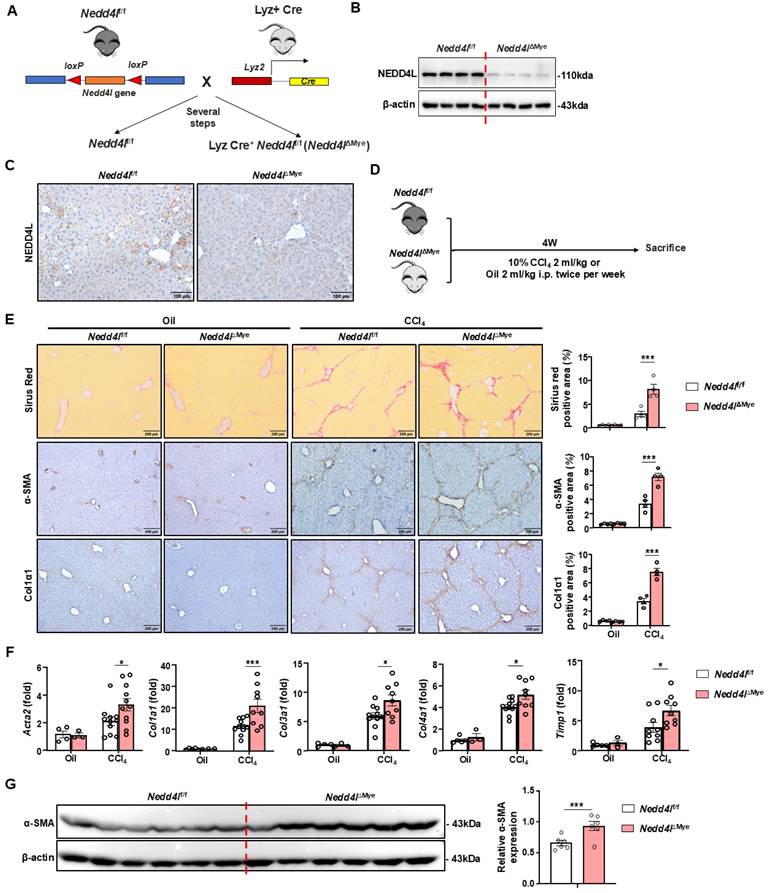
To clarify the role of NEDD4L in liver fibrosis, we constructed myeloid cell-specific Nedd4l knockout mice (Nedd4lΔMye) (Fig. 2A). The knockout efficiency was validated using Western blot by isolating bone marrow-derived macrophages (Fig. 2B) and immunofluorescence (Fig. 2C, Supporting Fig. S1B), confirming significantly reduced NEDD4L expression in macrophages compared to littermate controls. To induce liver fibrosis, both Nedd4lΔMye and their Nedd4lfl/fl counterparts were subjected to a 4-week regimen of intraperitoneal injections of CCl4 or olive oil (vehicle control) (Fig. 2D). Myeloid cell-specific Nedd4l deficiency exacerbated liver fibrosis as demonstrated that Nedd4lΔMye mice had greater collagen deposition and elevated α-smooth muscle actin (α-SMA) (Fig. 2E). While no significant fibrotic changes or intergroup differences were observed in the vehicle-treated mice, CCl4-treated Nedd4lΔMye mice displayed substantially expanded fibrotic areas. Consistent with these morphological findings, RT-qPCR analysis further demonstrated that hepatic expression of several key profibrogenic genes including Acta2, Col1a1, Col3a1, Col4a1, and Timp1, was significantly elevated in Nedd4lf/f mice compared to vehicle controls (Fig. 2F). Subsequent western blot validation confirmed a robust elevation in α-SMA protein expression in CCl4-treated Nedd4lΔMye mice compared to that in Nedd4lf/f counterparts (Fig. 2G). Collectively, these data indicate that myeloid cell-specific deletion of Nedd4l aggravates CCl4-induced experimental mouse liver fibrosis.
Myeloid cell-specific knockout of Nedd4l exacerbates CCl4-induced liver fibrosis. (A) Strategy for generating myeloid cell-specific Nedd4l knockout mice. (B-C) Validation of Nedd4l knockout efficiency by western blot in BMDMs and by IHC staining in liver sections. (D) Eight-week-old male Nedd4lΔMye mice and their Nedd4lf/f littermate controls were subjected to CCl4 or olive oil injection for 4 weeks. (E) Representative images of Sirius red staining (Scale bar: 200 μm), α-SMA staining (Scale bar: 200 μm) and Col1α1 staining (Scale bar: 200 μm) from Nedd4lf/f and Nedd4lΔMye mice are shown. The percentage of positive area was quantified. (F) The hepatic expression of Acta2, Col1a1, Col3a1, Col4a1 and Timp1 were analyzed by RT-qPCR in liver tissues from Nedd4lf/f and Nedd4lΔMye mice. (G) Protein levels of α-SMA in liver tissues were assessed by Western blotting. β-actin was used as a loading control. Values represent mean ± SEM. *p<0.05, ***p<0.001.
Myeloid cell-specific knockout of Nedd4l worsens CDAHFD-induced liver fibrosis
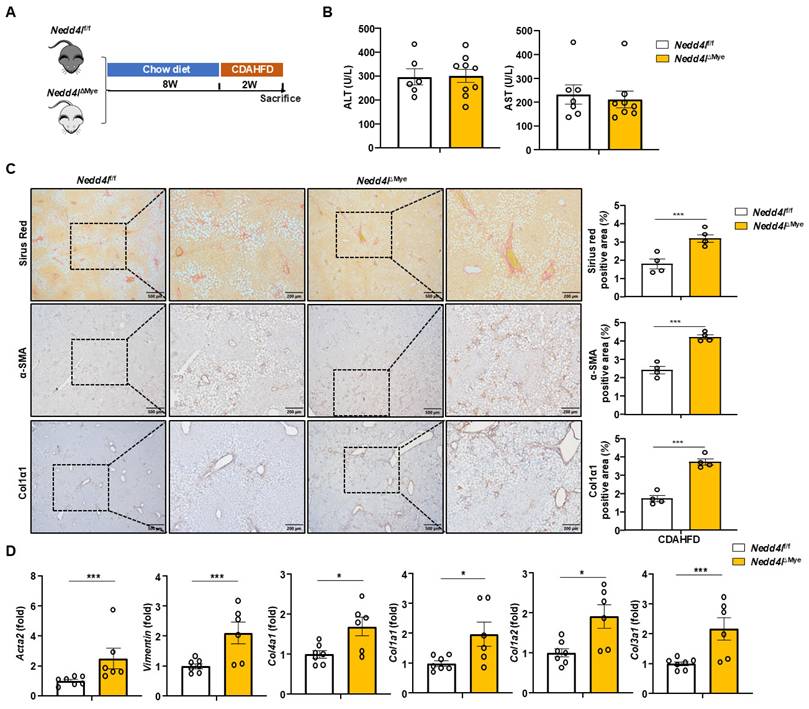
To define the role of macrophage NEDD4L in nutrition-induced hepatic fibrosis, we employed a well-established dietary model using a CDAHFD-induced liver fibrosis model, which recapitulates key aspects of metabolic dysfunction-associated steatohepatitis (MASH) and progressive fibrogenesis [25] (Fig. 3A). Following two weeks of CDAHFD feeding, serum ALT and AST levels were comparable between Nedd4lΔMye mice and their Nedd4lf/f littermate controls (Fig. 3B), suggesting comparable initial liver damage under this short-term nutritional challenge. Interestingly, Nedd4lΔMye mice exhibited substantially expanded fibrotic areas along with significantly intensified deposition of α-SMA⁺ and Col1α1⁺ cells (Fig. 3C). RT-qPCR profiling demonstrated remarkable upregulation of a panel of fibrogenic genes in the livers of Nedd4lΔMye mice including Acta2, Col1a1, Col3a1, Col4a1, and Vim (Fig. 3D), indicating myeloid cell-specific Nedd4l deficiency also exacerbated CDAHFD-induced liver fibrosis. The accelerated fibrosis in Nedd4lΔMye mice after only two weeks of CDAHFD feeding suggested a critical role for macrophage NEDD4L in restraining the early fibrogenic response. To validate this function in a distinct, rapid model of monocyte-driven fibrosis, we employed bile duct ligation (BDL) model (Supporting Fig. S2A). Immunofluorescence analysis confirmed NEDD4L expression in liver macrophages following BDL (Supporting Fig. S2B). Consistent with our previous findings, Nedd4lΔMye mice developed greater liver fibrosis after BDL compared to Nedd4lf/f littermates as evidenced by increased collagen deposition (Supporting Fig. S2C), and by elevated mRNA expression of several key fibrotic markers (e.g., Col1a1, Acta2) as well as pro-fibrogenic genes in macrophages (e.g., Fabp5, Gpnmb) (Supporting Fig. S2D), underscoring the generalizable role of NEDD4L in suppressing the fibrogenic activity of macrophages. The consistent exacerbation across both nutritional and toxic injury paradigms underscores a central protective role for macrophage NEDD4L in restraining liver fibrogenesis, independent of the original insult, and highlights its potential as a cross-disease modulator of liver fibrosis.
Nedd4l deficiency in macrophages aggravates CDAHFD-induced liver fibrosis. (A) Eight-week-old male Nedd4lΔMye mice and their littermate Nedd4lfl/f mice were fed CDAHFD for 2 weeks to establish the experimental MASH-related liver fibrosis model. (B) Serum ALT and AST levels were measured. (C) Representative images of Sirius red staining (Scale bar: 200 μm), α-SMA staining (Scale bar: 200 μm) and Col1α1 staining (Scale bar: 200 μm) from Nedd4lf/f and Nedd4lΔMye mice are shown. The percentage of positive area was quantified. (D) The hepatic expression of Acta2, Vimentin, Col4a1, Col1a1, Col1a2 and Col3a1 were analyzed by RT-qPCR in liver tissues from Nedd4lf/f and Nedd4lΔMye mice. Values represent mean ± SEM. *p<0.05, ***p<0.001.
Myeloid cell-specific knockout of Nedd4l promotes scar-associated macrophage (SAM) differentiation in liver fibrosis
Extensive evidence indicate that macrophages contribute to liver fibrosis through diverse mechanisms. They are highly plastic and can adopt various activation states, broadly categorized into two major phenotypes: classically activated (M1) pro-inflammatory macrophages and alternatively activated (M2) macrophages, which are often associated with tissue repair and pro-fibrotic responses [26]. To understand whether immune cell infiltration was involved in exacerbated liver fibrosis in Nedd4lΔMye mice, we performed immunohistochemical staining of F4/80, MPO and CD3. Unexpectedly no significant differences in the number of F4/80⁺ macrophages, MPO⁺ neutrophils, and CD3⁺ T cells were observed between Nedd4lΔMye and Nedd4lf/f mice (Supporting Fig. S3A). RT-qPCR corroborated these findings: although CCl4 challenge significantly upregulated several key inflammatory genes including Il1b, Il6, Tnfa, Mmp1, Cxcl1, and Ccl2 in Nedd4lf/f mice, hepatic expression of these genes did not differ significantly between Nedd4lΔMye and Nedd4lf/f groups (Supporting Fig. S3B). Serum ALT and AST levels were similarly elevated following CCl₄ administration, without significant differences between Nedd4lΔMye and Nedd4lf/f mice, and baseline levels of serum ALT and AST were also comparable between untreated groups, indicating that myeloid-specific deletion of Nedd4l does not cause spontaneous hepatocellular injury (Supporting Fig. S3C).This pattern was consistently observed in the CDAHFD nutritional model as well, with no intergroup differences in F4/80⁺ or MPO⁺ immune cell densities (Supporting Fig. S4A) or in the expression of inflammatory genes such as Il1b, Tnfa, and Ccl2 (Supporting Fig. S4B). These findings clearly demonstrated that myeloid cell-specific deletion of Nedd4l exacerbates liver fibrosis possibly through inflammation-independent mechanisms.
Given the absence of a discernible inflammatory effect, we turned our attention to another macrophage subset critically involved in liver fibrogenesis: pro-fibrotic Trem2+FABP5+SPP1+CD9+ scar-associated macrophages (SAMs), which have been implicated in hepatic injury repair and activation of hepatic stellate cells through production of TGF-β1—a process that, when dysregulated, directly exacerbates fibrosis[6, 27]. Single-cell transcriptomic analysis of human fibrotic livers revealed that SAMs exhibited markedly elevated Nedd4l expression compared to negligible levels in healthy controls (https://singlecell.broadinstitute.org/single_cell) (Fig. 4A-B). Furthermore, these fibrotic SAMs concurrently highly expressed pro-fibrotic mediators including CCL22, VEGFA, and TLR1 (Supporting Fig. S5A). More importantly, immunofluorescence staining and quantitative cell counting revealed a significant increase in FABP5⁺, a specific marker for SAMs, in Nedd4lΔMye livers compared to controls (Fig. 4C). Consistent with this finding, co-staining for IBA1⁺CD206⁺ macrophages similarly revealed a significant expansion in this population (Supporting Fig. S5B), suggesting an overall increase in the SAM-profibrogenic macrophage compartment in Nedd4lΔMye mice. RT-qPCR analysis further confirmed upregulation of SAM signature genes including Spp1, Cd63, Gpnmb, and Fabp5 as well as pro-fibrotic mediators including Mrc1, Chil3 (Ym1), Tlr8, and Tgfb1 in CCl4-treated Nedd4lΔMye mice (Fig. 4D). In addition, the expression of pro-fibrotic macrophage markers (TLR1 and CD206) was greater in Nedd4lΔMye mice than Nedd4lf/f controls after CCl4 injection (Fig. 4E). These results suggested that Nedd4l deficiency in macrophages promotes SAM differentiation, thereby accelerating toxin-induced liver fibrosis progression.
Myeloid cell-specific knockout of Nedd4l promotes scar-associated macrophage (SAM) differentiation during liver fibrosis. (A) Analysis of Nedd4l expression in liver macrophages from patients with liver fibrosis by analyzing single-cell sequencing database (Liver Cell Atlas: https://singlecell.broadinstitute.org). (B) Analysis of Nedd4l expression in SAMs from patients with liver fibrosis by analyzing liver single-cell sequencing database (Liver Cell Atlas: https://singlecell.broadinstitute.org). (C) Representative immunofluorescence images of IBA1 (red), FABP5 (green), and DAPI (white) (Scale bar: 10 μm) in the livers of CCl4-treated Nedd4lf/f and Nedd4lΔMye mice. The number of positive cells was quantified. (D) The hepatic expression of SAM-related profibrotic genes were analyzed by RT-qPCR in liver tissues from oil or CCl4-treated Nedd4lf/f and Nedd4lΔMye mice. (E) Representative images of CD163 staining (Scale bar: 100 μm) and TLR1 staining (Scale bar: 200 μm) from CCl4-treated Nedd4lf/f and Nedd4lΔMye mice are shown. The number of positive cells was quantified. (F) Representative immunofluorescence images of IBA1 (red), FABP5 (green), and DAPI (white) (Scale bar: 10 μm) in the livers of CDAHFD-fed Nedd4lf/f and Nedd4lΔMye mice are shown. The number of positive cells was quantified. (G) The hepatic expression of SAM-related profibrotic genes were analyzed by RT-qPCR in liver tissues from CDAHFD-fed Nedd4lf/f and Nedd4lΔMye mice. (H) Representative images of FABP5 staining (Scale bar: 100 μm), TREM2 staining (Scale bar: 100 μm), CD9 staining (Scale bar: 100 μm), and CD206 staining (Scale bar: 100 μm) from CDAHFD-fed Nedd4lf/f and Nedd4lΔMye mice are shown. The number of positive cells was quantified. Values represent mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
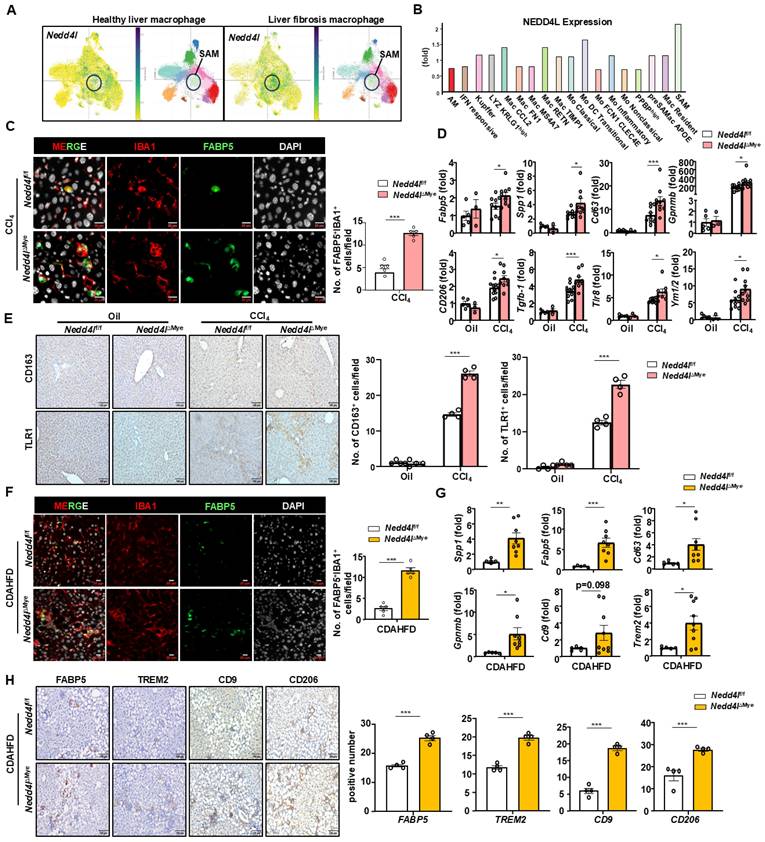
Consistent with the findings from the CCl4-induced liver fibrosis model, immunofluorescence staining of murine fibrotic livers also showed elevated IBA1⁺FABP5⁺ macrophages in Nedd4lΔMye mice compared to Nedd4lf/f controls after CDAHFD feeding (Fig. 4F). Subsequent RT-qPCR analysis corroborated these results, revealing pronounced upregulation of SAM-associated transcripts including Spp1, Cd63, Trem2, Gpnmb, and Fabp5, in Nedd4lΔMye livers with Cd9 showing a consistent upward trend (Fig. 4G). Immunohistochemical analysis of liver sections from mice fed CDAHFD for two weeks showed significantly elevated expression of SAM markers (TREM2, CD9, FABP5) and pro-fibrotic macrophage marker CD206 in Nedd4lΔMye mice compared to Nedd4lfl/fl controls (Fig. 4H). Notably, the pro-fibrotic polarization of Nedd4l-deficient macrophages was not attributable to altered cell survival or a generalized hyper-inflammatory state, as their viability and classic inflammatory response to lipopolysaccharide (LPS) remained comparable to controls (Supporting Fig. S6A). Taken together, these data from both toxin-induced and nutrition-based fibrotic models demonstrate that myeloid cell-specific knockout of Nedd4l potentiates SAM differentiation and pro-fibrotic activation independently of classical inflammatory pathways.
NEDD4L limits pro-fibrotic macrophage phenotype via regulation of SMAD3 phosphorylation
To elucidate the mechanisms underlying post-TGF-β1 responses in macrophages, we first performed protein-protein interaction analysis (https://cn.string-db.org), which identified NEDD4L as a key regulator of SMAD3, suggesting a potential role in modulating TGF-β1 signaling activity (Fig. 5A). As an E3 ubiquitin ligase, NEDD4L primarily exerts its biological functions by regulating target proteins through ubiquitination. Computational structural analysis of ubiquitin ligase-substrate interactions predicted that NEDD4L directly bound phosphorylated SMAD3 (https://www.rcsb.org) (Fig. 5B). Subsequent molecular profiling demonstrated that myeloid cell-specific Nedd4l knockout significantly potentiates SMAD2/3 activation as evidenced that TGF-β1 stimulation resulted in markedly enhanced phosphorylation of SMAD2/3 (p-SMAD2/3) in macrophages from Nedd4lΔMye mice compared to that from Nedd4lf/f controls, while total SMAD2/3 protein levels remained unchanged (Fig. 5C). Importantly, this effect was specific to the canonical TGF-β pathway, as non-canonical pathways including ERK and p38 MAPK signaling were unaltered (Fig. 5C). Importantly, this enhanced TGF-β1 responsiveness in Nedd4l-deficient macrophages was specific, as their viability and classical inflammatory response to LPS treatment were unaltered (Supporting Fig. S6B).
NEDD4L limits pro-fibrotic macrophage polarization via inhibiting SMAD3 phosphorylation. (A) Protein interaction network analysis (https://cn.string-db.org). (B) Structural prediction of the interaction between NEDD4L and phosphorylated SMAD3 protein (https://www.rcsb.org/). (C) BMDMs isolated from Nedd4lf/f and Nedd4lΔMye mice were stimulated with M-CSF (20 ng/ml) for 7 days, and then stimulated with 10 ng/ml TGF-β1. Western blot analysis of TGF-β1 signal pathway. β-actin was used as a loading control. (D) Representative images of p-Smad3 staining (Scale bar: 50 μm) from oil-treated, CCl4- treated and CDAHFD-fed Nedd4lf/f and Nedd4lΔMye mice are shown. The number of positive cells was quantified. (E) Representative images of p-Smad3 and IBA1 (Scale bar: 50 μm) double staining of liver tissue sections from CCl4-treatedand CDAHFD-fed Nedd4lf/f and Nedd4lΔMye mice are shown. The number of positive cells was quantified. (F) Schematic diagram of experimental design. BMDMs isolated from C57BL/6J mice were differentiated with M-CSF (20 ng/ml, 7 days) prior to a 24-hour stimulation with TGF-β1 (10 ng/ml), with or without SIS3 (5 μM). Conditional BMDM medium were co-cultured with LX-2 cells. (G) The expression of fibrogenic genes were analyzed by RT-qPCR in LX-2 cells after culture with conditional medium from BMDMs. (H) Schematic diagram of experimental design. BMDMs isolated from Nedd4lf/f and Nedd4lΔMye mice were differentiated with M-CSF (20 ng/ml, 7 days) prior to a 24-hour stimulation with TGF-β1 (10 ng/ml), with or without SIS3 (5 μM). (I) The expression of pro-fibrotic genes was analyzed by RT-qPCR in BMDMs after culture with different conditional medium. Values represent mean ± SEM. *p< 0.05, ***p < 0.001.
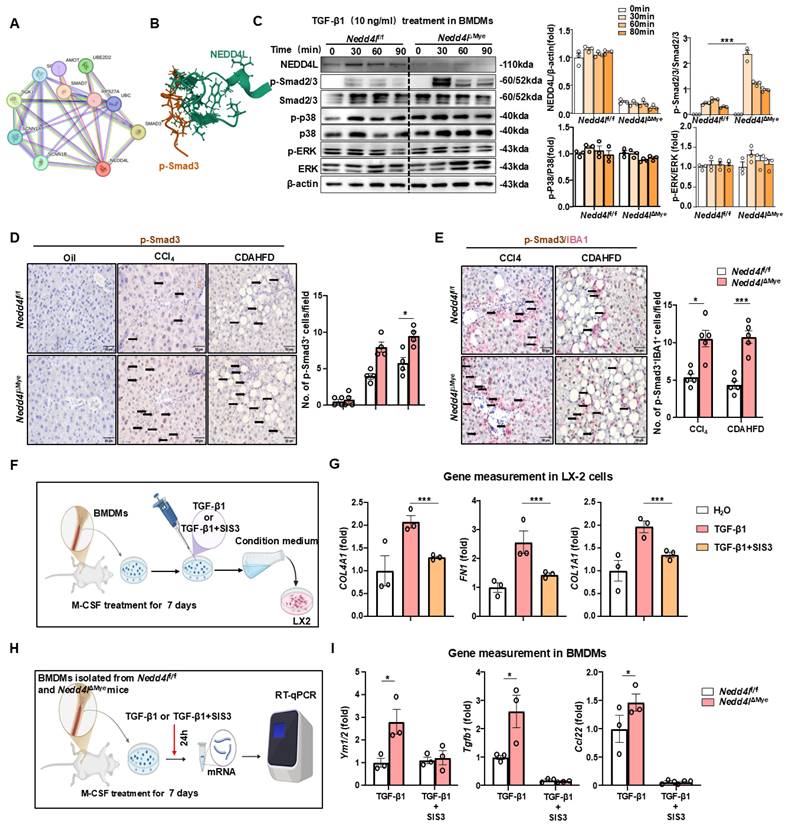
Next, immunohistochemistry of liver sections revealed a substantially more prominent infiltration of p-SMAD3⁺ immune cells in fibrotic livers than that in control tissues (Fig. 5D). To this end, immunohistochemical double staining was performed. A notably expanded population of IBA1⁺/p-SMAD3⁺ macrophages was readily detectable in Nedd4lΔMye fibrotic livers compared to Nedd4lf/f controls (Fig. 5E), these observed increases were confirmed by quantitative analysis, providing histological evidence that Nedd4l deficiency in macrophages enhances SMAD3 activation specifically in macrophages under fibrotic conditions.
To investigate the contribution of macrophage p-SMAD3 to HSC activation, we collected conditioned medium from the pharmacological SMAD3 inhibitor SIS3-treated macrophages (5mM refreshed every 12h for 48h) primary bone marrow-derived macrophages (BMDMs) (Fig. 5F). This conditioned medium significantly attenuated the activation of LX-2 cells as indicated by reduced expression of TGFB1, FN1 and COL4A1 (Fig. 5G). Pharmacological inhibition of p-SMAD3 with SIS3 obviously diminished the pro-fibrotic gene signature (Ym1/2, Tgfb1, Ccl22) in BMDMs from Nedd4lΔMye mice, thereby functionally establishing the dependence of this enhanced fibrogenic phenotype on p-SMAD3 signaling (Fig. 5H-I). These comprehensive data suggest that Nedd4l deficiency in macrophages drives pro-fibrotic polarization primarily through enhanced p-SMAD3-dependent signaling mechanisms, which in turn promotes HSC activation and contributes to fibrosis pathogenesis.
Given that TGF-β1 participates in pro-fibrotic macrophage differentiation [28], we sought to further elucidate the mechanistic impact of macrophage Nedd4l deficiency on this process through a comprehensive in vitro approach. Primary BMDMs were differentiated and employed to model physiological conditions in vitro. BMDMs were differentiated with M-CSF for 7 days to attain a mature macrophage phenotype, followed by TGF-β1 stimulation for 24 hours (Fig. 6A). RT-qPCR analysis demonstrated that under basal conditions, the expression of SAM macrophage-related genes (Trem2, Cd9, and Cd63) and pro-fibrogenic genes (Arg1, Ccl2, and Vegfa) did not differ significantly between unstimulated BMDMs from Nedd4lf/f and BMDMs from Nedd4lΔMye. However, upon TGF-β1 stimulation for 24 hours, BMDMs from Nedd4lΔMye mice exhibited a markedly greater amplitude of induction in the expression of these genes compared to that from Nedd4lfl/fl controls (Fig. 6B). These results indicate that Nedd4l deficiency in macrophages predisposes macrophages toward a pro-fibrogenic SAM phenotype.
Myeloid cell-specific knockout of Nedd4l enhances TGF-β1-induced pro-fibrotic macrophage polarization in vitro. (A) Schematic diagram of experimental design. BMDMs isolated from Nedd4lf/f and Nedd4lΔMye mice were stimulated with M-CSF (20 ng/ml) 7 days, and then stimulated with 10 ng/ml TGF-β1 for 24 hours. (B) The expression of pro-fibrotic genes was analyzed by RT-qPCR in BMDMs. (C) Schematic diagram of experimental design. BMDMs isolated from Nedd4lf/f and Nedd4lΔMye mice were stimulated with M-CSF (20 ng/ml) 7days, and then stimulated with 10 ng/ml TGF-β1 for 24 hours. Conditional BMDM medium were co-cultured with LX-2 cells. (D) The expression of fibrogenic genes was analyzed by RT-qPCR in LX-2 cells treated with conditional medium from Nedd4lf/f and Nedd4lΔMye macrophages. (E) Protein levels of Col1α1 in LX-2 cells treated with conditional medium from Nedd4lf/f and Nedd4lΔMye macrophages were assessed by Western blotting. β-actin was used as a loading control. Values represent mean ± SEM. *p< 0.05, ***p< 0.001.
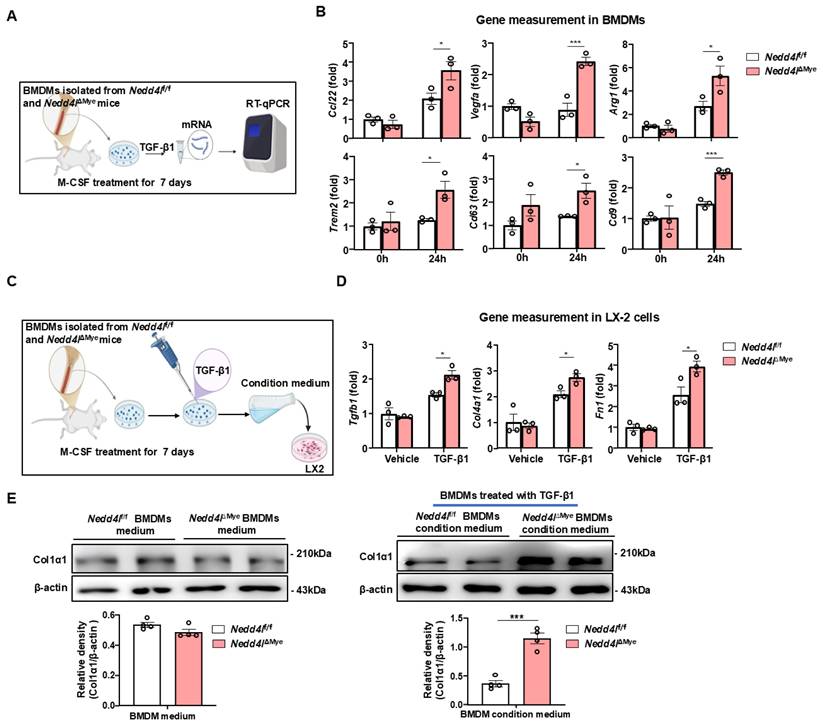
To determine whether the secretory phenotype of Nedd4l-deficient macrophages functionally activated HSCs, we established a simplified co-culture system. Unactivated LX-2 human HSCs were treated with conditioned medium collected from BMDMs, with or without exogenous TGF-β1 supplementation (Fig. 6C). LX-2 cells exposed to conditioned medium from Nedd4lΔMye BMDMs in the presence of TGF-β1 exhibited significantly higher expression of several key fibrogenic genes, including TGFB1, COL4A1, and FN1 (Fig. 6D). Western blot analysis further confirmed elevated protein levels of Col1α1 in LX-2 cells incubated with Nedd4lΔMye macrophage-conditioned medium (Fig. 6E), providing the compelling evidence that TGF-β1-treated Nedd4l deficient macrophages are more prone to promote HSC activation compared with TGF-β1-treated control macrophages. Collectively, these in vitro data suggest that Nedd4l deficiency in macrophages not only intrinsically enhances their pro-fibrotic transcriptional response to TGF-β1 but also augments their capacity to activate HSCs via paracrine mechanisms. To further validate these findings and address potential species differences, we performed the key co-culture experiments using the murine HSC cell line JS-1 cells. Conditioned medium from Nedd4lΔMye BMDMs similarly enhanced the expression of fibrogenic genes (Acta2, Col1a1) in JS-1 cells, corroborating our results in a species-matched system (Supporting Fig. S7).
NEDD4L mediates ubiquitination of phosphorylated SMAD3, leading to its degradation
To investigate whether NEDD4L directly ubiquitinates phosphorylated SMAD2/3, we conducted a series of well-controlled experiments in HEK293T cell lines. First, we transfected HEK293T cells with a NEDD4L-Flag overexpression plasmid, stimulated them with TGF-β1 for 1 hour, and analyzed the cell lysates by Western blot. As shown in Fig. 7A, NEDD4L overexpression significantly reduced the protein levels of phosphorylated SMAD2/3 (p-SMAD2/3) without altering total SMAD2/3 expression, suggesting a specific regulatory effect on the activated form of these transcription factors. To determine whether this reduction was mediated through ubiquitination-dependent proteasomal degradation, we performed TGF-β1 stimulation in the presence of MG132, a potent proteasome inhibitor. MG132 treatment effectively rescued the decrease in phosphorylated SMAD2/3 levels induced by NEDD4L overexpression (Fig. 7B), supporting that NEDD4L promotes proteasomal degradation of p-SMAD2/3.
NEDD4L promotes ubiquitination of phosphorylated SMAD3, leading to its degradation. (A) HEK293T cells were transfected with NEDD4L overexpression or control plasmid for 24 hours prior to TGF-β1 stimulation. Protein levels were assessed by Western blotting. (B) HEK293T cells were transfected with NEDD4L or control plasmid for 24 hours prior to a 1-hour TGF-β1 stimulation with or without MG132. Protein levels were assessed by Western blotting. (C) Co-immunoprecipitation of Myc-Smad3 with Flag-NEDD4L from the transfected HEK293T cell lysates using an anti-Flag antibody, followed by western blot analyses. (D) HEK293T cells were co-transfected with HA-tagged ubiquitin, Flag-Nedd4L, and Myc-Smad3. Myc immunoprecipitates from whole-cell lysates were analyzed by immunoblotting with the indicated antibodies. (E) HEK293T cells were transfected with NEDD4L or control siRNA for 24 hours prior to HA-ubiquitin plasmid transfection. After another 24 hours, the cells were stimulated with TGF-β1 for 1 hour. Smad2/3 immunoprecipitates and whole-cell lysates were then analyzed by immunoblotting. (F) Liver tissues of patients with different stage of liver fibrosis were collected. Representative images of NEDD4L staining (Scale bar: 200 μm and 50 μm) are shown. (G) Schematic model of NEDD4L-p-SMAD3 axis in macrophages during liver fibrosis. TGF-β1 signaling initiates pro-fibrotic response in macrophages by inducing p-SMAD3-mediated SAM differentiation. Concurrently, NEDD4L elevation in macrophages forms a negative feedback circuit to limit liver fibrosis by promoting p-SMAD3 degradation. Values represent mean ± SEM. ***p<0.001.
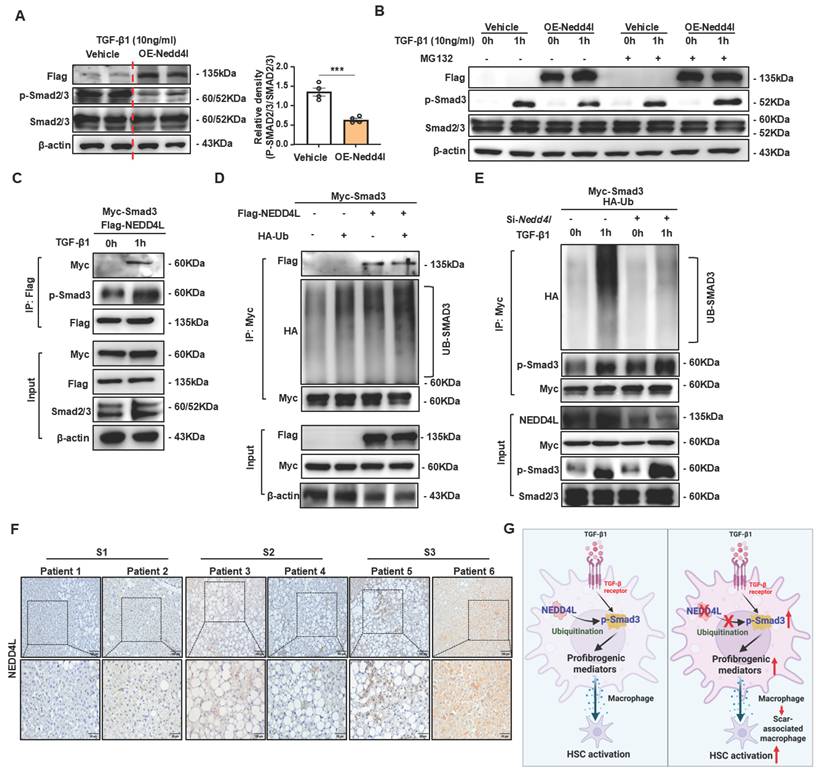
We next examined whether NEDD4L directly interacted with p-SMAD3. HEK293T cells were co-transfected with SMAD3-Myc and NEDD4L-Flag plasmids, stimulated with TGF-β1, and then subjected to co-immunoprecipitation (co-IP) using Flag antibodies. As shown in Fig. 7C, p-SMAD3-Myc was specifically co-precipitated with NEDD4L-Flag only upon TGF-β1 stimulation. Critically, p-SMAD2/3 was detected in these immunoprecipitates, confirming that NEDD4L binds directly to p-SMAD3 in a TGFβ-dependent manner.
To further evaluate whether this interaction leads to ubiquitination of p-SMAD3, we co-transfected HEK293T cells with Ub-HA and SMAD3-Myc, with or without NEDD4L-Flag. Following TGF-β1 stimulation and immunoprecipitation of SMAD3-Myc, we observed significantly enhanced ubiquitination of p-SMAD3 in the presence of NEDD4L (Fig. 7D). Conversely, when endogenous NEDD4L was knocked down using shRNA targeting NEDD4L (shNEDD4L), TGF-β1-induced ubiquitination of p-SMAD3 was markedly attenuated (Fig. 7E), providing additional genetic evidence for the essential role of NEDD4L in this process. Taken together, these results demonstrate that NEDD4L directly binds to phosphorylated SMAD3 upon TGFβ stimulation and promotes its ubiquitination and subsequent proteasomal degradation. This mechanism maintains tight control over the duration and intensity of TGFβ signaling.
To investigate the clinical correlation and significance of NEDD4L, we assembled a cohort of human liver tissues from patients with fibrosis of multiple etiologies and across a spectrum of disease severity. Immunohistochemistry staining of NEDD4L showed that NEDD4L protein expression was elevated in human severe fibrotic liver tissues compared with mild fibrotic tissues (Fig. 7F).
Discussion
NEDD4L, a member of the NEDD4 family of E3 ubiquitin ligases, is known to regulate protein degradation and affect various cellular processes, including TGF-β signaling [22]. However, its cell type-specific functions, particularly in macrophages during the development of liver fibrosis, remain poorly understood. In this study, we demonstrate that myeloid cell-specific deletion of Nedd4l exacerbates liver fibrosis by promoting SAM-related macrophage differentiation via enhancing TGF-β/SMAD3 signaling in macrophages. These findings uncover a critical role for macrophage NEDD4L in restraining liver fibrogenesis and highlight its potential as a therapeutic target for the treatment of liver fibrosis (Fig. 7G).
Although NEDD4L has been implicated in various pathological contexts, including viral infection [23], brain injury [24], organ fibrosis [29, 30], and cancers [31, 32], its function in liver fibrosis—particularly within macrophages—has not been previously elucidated. A key mechanistic insight from our study is the identification of p-SMAD3 as a direct substrate of NEDD4L in macrophages. Previous reports indicated that NEDD4L can ubiquitinate SMAD2/3 and TGF-β receptors [33], but its role in modulating SMAD3 stability in macrophages was unknown. We showed that TGF-β1 stimulation promoted the physical interaction between NEDD4L and p-SMAD3, leading to its ubiquitination and degradation. Crucially, NEDD4L specifically targets activated, not resting-state, Smad2/3 for degradation. This exquisite selectivity requires a two-step verification: first, TGF-β1-induced activation, and second, CDK8/9-mediated phosphorylation of a specific pT-PY motif that is recognized by the WW2 domain of NEDD4L. This specific mechanism ensures precise termination of TGF-β signaling without disturbing the basal cellular pool of Smad2/3 [22]. In the absence of NEDD4L, p-SMAD3 accumulates and drives sustained expression of pro-fibrotic genes, thereby accelerating fibrosis. This places NEDD4L as a central negative regulator of TGF-β signaling in macrophages, operating at the level of downstream SMAD activation. Notably, the profibrotic effect of myeloid NEDD4L deficiency was not primarily mediated by amplifying classic inflammation, as evidenced by unaltered or even reduced expression of cytokines including Il6 and Tnfa in fibrotic Nedd4lΔMye livers. This indicates that NEDD4L specifically restrains a fibrogenesis-prone macrophages, which can be dissociated from the canonical pro-inflammatory response. Although NEDD4L has been reported to ubiquitinate SMAD2 in certain contexts [34], our results also revealed that Nedd4l deficiency attenuates the ubiquitin-mediated degradation of p-SMAD2, leading to its accumulation in our liver fibrosis models (data not shown). However, as previous studies suggest that SMAD2 has a minimal impact on macrophage differentiation [35]. The potential non-canonical roles of SMAD2 in macrophages present an intriguing subject for future research. Our data indicate that its predominant fibro-suppressive function in hepatic macrophages is mediated through SMAD3. While both SMAD2 and SMAD3 are key effectors of TGF-β1 signaling, they often play non-redundant roles, with SMAD3 being particularly critical in driving pro-fibrotic gene expression. It is worth noting that the basal turnover of p-SMAD3 is primarily mediated by the ubiquitin-proteasome system, with several E3 ligases—including SMURF1/2 [33, 34]—implicated in this process. The classical negative feedback regulator SMAD7 [36], which is induced by TGF-β signaling, is known to recruit SMURF E3 ligases to target both TGF-β receptors and activated SMAD2/3 complexes for degradation. In contrast, our data suggest that NEDD4L exhibits more selective activity towards p-SMAD3 in macrophages, potentially operating independently of SMAD7-mediated recruitment. This specificity is therapeutically crucial, as global inhibition of SMAD degradation through central regulators like SMAD7 may cause widespread unintended consequences, whereas targeting the more discrete NEDD4L-pSMAD3 interaction offers a unique opportunity for cell-type and substrate-selective intervention in fibrotic diseases.
A critical question arising from our findings is how the accumulation of p-SMAD3, resulting from Nedd4l deletion in macrophages, precisely orchestrates the phenotypic transition of macrophages towards a pro-fibrotic SAM state. We hypothesized that sustained SMAD3 activation initiates a transcriptional reprogramming that extends beyond classical TGF-β1 targets. Specifically, the prolonged nuclear retention of p-SMAD3 may directly bind to and activate promoters of key pro-fibrotic mediators, creating an autocrine loop that perpetuates the activated state of SAMs. Furthermore, p-SMAD3 likely collaborates with other transcription factors and epigenetic modifiers to establish a stable pro-fibrotic gene expression signature. Supporting this notion, the anti-fibrotic drug pirfenidone (PFD) has been shown to ameliorate fibrosis in radiation-induced lung injury (RILI) primarily by suppressing p-Smad3, which subsequently attenuates M2 macrophage infiltration. This inhibition subsequently attenuates M2 macrophage infiltration and disrupts the downstream activation of the TGF-β1/Smad3 pathway [37]. Through these mechanisms, SAMs are known to actively promote hepatic fibrogenesis by sustaining TGF-β secretion, producing pro-fibrotic mediators such as PDGF and CTGF, directly activating hepatic stellate cells, and inhibiting ECM degradation—collectively driving the progression of liver fibrosis [38, 39]. The exact repertoire of genes directly regulated by the NEDD4L-p-SMAD3 axis in SAMs and its crosstalk with other signaling pathways represents a vital area for future investigation, as it will provide deeper insights into the molecular drivers of macrophage plasticity in fibrosis.
Another significant finding is the role of NEDD4L in restraining SAM differentiation. SAMs have recently emerged as key contributors to fibrosis progression [39]. Our data show that Nedd4l deficiency in macrophages drives SAM expansion and enhances their pro-fibrotic output, suggesting that NEDD4L acts as an intrinsic brake on macrophage phenotypic switching toward a profibrotic state. From a clinical perspective, targeting NEDD4L may offer a novel therapeutic strategy for liver fibrosis. Unlike broad TGF-β inhibitors [40], which often cause systemic side effects such as immune suppression or cardiovascular complications, enhancing NEDD4L activity could provide a more specific means of dampening fibrogenic signaling within the liver microenvironment. Mechanistically, the role of NEDD4L may extend beyond SMAD3 regulation. As an E3 ubiquitin ligase, NEDD4L likely has multiple substrates in macrophages that contribute to its anti-fibrotic function. For example, NEDD4L is known to regulate membrane receptors and transporters that could influence macrophage migration, cytokine secretion, or cross-talk with other hepatic cells [33]. Additionally, the WW domains of NEDD4L mediate interactions with proline-rich motifs present in many signaling proteins [8], suggesting possible involvement in other pathways such as Wnt/β-catenin [41] or Notch [42], which are also implicated in fibrogenesis. The specific upstream regulators of NEDD4L in fibrotic macrophages—such as cytokines, metabolic signals, or mechanical stress—also warrant further investigation, as they may reveal additional targets for intervention.
In conclusion, our study uncovers macrophage NEDD4L as a crucial regulator of liver fibrosis through ubiquitin-mediated degradation of p-SMAD3 and suppression of SAM differentiation. These findings not only advance our understanding of the ubiquitin-proteasome system in immune-mediated fibrogenesis but also identify NEDD4L as a potential target for anti-fibrotic therapy.
Abbreviations
Acta2: Actin Alpha 2, Smooth Muscle; ALT: Alanine Aminotransferase; ARG1/Arg1: Arginase 1; AST: Aspartate Aminotransferase; BMDM: Bone Marrow-Derived Macrophage; CCl4: Carbon Tetrachloride; CCL2: C-C Motif Chemokine Ligand 2; CCL22: C-C Motif Chemokine Ligand 22; CDAHFD: Choline-deficient, L-amino acid-defined, High-fat Diet; Col1α1: Collagen Type I Alpha 1 Chain; Col3α1: Collagen Type III Alpha 1 Chain; Col4α1: Collagen Type IV Alpha 1 Chain; Co-IP: Co-Immunoprecipitation; CXCL1: C-X-C Motif Chemokine Ligand 1; DAPI: 4′,6-Diamidino-2-Phenylindole; ECM: Extracellular Matrix; ERK: Extracellular Signal-Regulated Kinase; FABP5/Fabp5: Fatty Acid Binding Protein 5; GPNMB: Glycoprotein Nmb; HECT: Homologous to the E6-AP Carboxyl Terminus; HSC: Hepatic Stellate Cell; IBA1: Ionized Calcium Binding Adaptor Molecule 1; IHC: Immunohistochemistry; IL1β: Interleukin 1 Beta; IL-6: Interleukin 6; M-CSF: Macrophage Colony-Stimulating Factor; MAPK: Mitogen-Activated Protein Kinase; MASH: Metabolic dysfunction-associated steatohepatitis; MASLD: Metabolic dysfunction-associated steatotic liver disease; MPO: Myeloperoxidase; MRC1: Mannose Receptor C-Type 1 (CD206); NEDD4L: Neural precursor cell expressed, developmentally down-regulated 4-like; p-ERK: Phosphorylated ERK; p-SMAD2/3: Phosphorylated SMAD2/3; p-p38: Phosphorylated p38 MAPK; PBS: Phosphate Buffered Saline; RT-qPCR: Quantitative Reverse Transcription Polymerase Chain Reaction; SAM: Scar-Associated Macrophage; scRNA-seq: Single-Cell RNA Sequencing; shRNA: Short Hairpin RNA; siRNA: Small Interfering RNA; SPP1/Spp1: Secreted Phosphoprotein 1 (Osteopontin); TGF-β1: Transforming Growth Factor Beta 1; TIMP1: Tissue Inhibitor of Metalloproteinases 1; TLR1/8: Toll-Like Receptor 1/8; TNFα: Tumor Necrosis Factor Alpha; TREM2: Triggering Receptor Expressed on Myeloid cells 2; VEGFA: Vascular Endothelial Growth Factor A; VIM: Vimentin.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was sponsored by the Noncommunicable Chronic Diseases-National Science and Technology Major Project (2024ZD0530604, China); the National Key Research and Development Program of China (2023YFA1800804, China); Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0830402); the National Natural Science Foundation of China (82270620, 82270601, China); Key Department Project of Huadong Hospital (2022, ZDXK2213).
Author contributions
YHH, SG, SL and SY designed and conducted the experiments and wrote the paper; FY, XL, SD, YC, ZZ, and YZ conducted some experiments and edited the manuscript; SH and SK edited the manuscript; PW collected human liver samples and supervised the project; YH and YWC supervised the whole project and the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastro Hepat. 2021;18:151-66
2. Yang Z, Liu Z, Liu W, Dong XC. Steatotic liver disease and cancer: from pathogenesis to therapeutic targets. eGastroenterology. 2025;3:e100218
3. Liu Y, Wang L. Extracellular vesicles targeting non-parenchymal cells: the therapeutical effect on liver fibrosis. eGastroenterology. 2024;2:e100040
4. Guilliams M, Bonnardel J, Haest B, Vanderborght B, Wagner C, Remmerie A. et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell. 2022;185:379-96 e38
5. Cho YE, Kwon YS, Hwang S. Heterogeneous population of macrophages in the development of non-alcoholic fatty liver disease. Liver Res. 2023;7:16-25
6. He Y, Chen Y, Qian S, van Der Merwe S, Dhar D, Brenner DA. et al. Immunopathogenic mechanisms and immunoregulatory therapies in MASLD. Cellular & Molecular Immunology. 2025
7. De Ponti FF, Bujko A, Liu Z, Collins PJ, Schuermans S, Maueroder C. et al. Spatially restricted and ontogenically distinct hepatic macrophages are required for tissue repair. Immunity. 2025;58:362-80.e10
8. Wang ZW, Hu XL, Ye MM, Lin M, Chu M, Shen X. NEDD4 E3 ligase: Functions and mechanism in human cancer. Semin Cancer Biol. 2020;67:92-101
9. Manning JA, Kumar S. Physiological Functions of Nedd4-2: Lessons from Knockout Mouse Models. Trends Biochem Sci. 2018;43:635-47
10. Lee DE, Yoo JE, Kim J, Kim S, Kim S, Lee H. et al. NEDD4L downregulates autophagy and cell growth by modulating ULK1 and a glutamine transporter. Cell Death Dis. 2020 11
11. Jiang L, Zhao J, Yang Q, Li M, Liu H, Xiao X. et al. Lysosomal-associated protein transmembrane 5 ameliorates non-alcoholic steatohepatitis by promoting the degradation of CDC42 in mice. Nat Commun. 2023;14:2654
12. Guo Q, Xin M, Lu Q, Feng D, Yang V, Peng LF. et al. A novel NEDD4L-TXNIP-CHOP axis in the pathogenesis of nonalcoholic steatohepatitis. Theranostics. 2023;13:2210-25
13. Yu Y, Hu S, Zhang T, Shi H, Li D, Huang Y. et al. Fas apoptotic inhibitor molecule 2 mitigates metabolic dysfunction-associated fatty liver disease through autophagic CRTC2 degradation. Exp Mol Med. 2025
14. Deng Z, Fan T, Xiao C, Tian H, Zheng Y, Li C. et al. TGF-beta signaling in health, disease, and therapeutics. Signal Transduct Target Ther. 2024;9:61
15. Shi YG, Massagué J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685-700
16. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastro Hepat. 2017;14:397-411
17. Massague J, Sheppard D. TGF-beta signaling in health and disease. Cell. 2023;186:4007-37
18. Viola MF, Chavero-Pieres M, Modave E, Delfini M, Stakenborg N, Estévez MC. et al. Dedicated macrophages organize and maintain the enteric nervous system. Nature. 2023;618:818-26
19. Yu XY, Buttgereit A, Lelios I, Utz SG, Cansever D, Becher B. et al. The Cytokine TGF-β Promotes the Development and Homeostasis of Alveolar Macrophages. Immunity. 2017;47:903 -+
20. Gunderson AJ, Yamazaki T, McCarty K, Fox N, Phillips M, Alice A. et al. TGFβ suppresses CD8 T cell expression of CXCR3 and tumor trafficking. Nat Commun. 2020 11
21. Kuratomi G, Komuro A, Goto K, Shinozaki M, Miyazawa K, Miyazono K. et al. NEDD4-2 (neural precursor cell expressed, developmentally down-regulated 4-2) negatively regulates TGF-ß (transforming growth factor-β) signalling by inducing ubiquitin-mediated degradation of Smad2 and TGF-β type I receptor. Biochem J. 2005;386:461-70
22. Gao S, Alarcón C, Sapkota G, Rahman S, Chen PY, Goerner N. et al. Ubiquitin Ligase Nedd4L Targets Activated Smad2/3 to Limit TGF-β Signaling. Mol Cell. 2009;36:457-68
23. Gao P, Ma X, Yuan M, Yi Y, Liu G, Wen M. et al. E3 ligase Nedd4l promotes antiviral innate immunity by catalyzing K29-linked cysteine ubiquitination of TRAF3. Nat Commun. 2021 12
24. Xia XH, Yang Z, Zhang JW, Fu XJ, Han B, Xiong QJ. et al. E3 ligase Nedd4L promotes macrophage M1 polarization and exacerbates brain damage by TRAF3/TBK1 signaling pathway after ICH in mice. Immunol Lett. 2023;264:36-45
25. Yang L, Li W, Chen Y, Ya R, Qian S, Liu L. et al. Downregulation of ubiquitous microRNA-320 in hepatocytes triggers RFX1-mediated FGF1 suppression to accelerate MASH progression. Acta Pharm Sin B. 2025;15:4096-114
26. Horn P, Tacke F. Metabolic reprogramming in liver fibrosis. Cell Metabolism. 2024 36
27. Wynn TA, Vannella KM. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity. 2016;44:450-62
28. Zhang F, Wang HS, Wang XF, Jiang GM, Liu H, Zhang G. et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget. 2016;7:52294-306
29. Manning JA, Shah SS, Nikolic A, Henshall TL, Khew-Goodall Y, Kumar S. The ubiquitin ligase NEDD4-2/NEDD4L regulates both sodium homeostasis and fibrotic signaling to prevent end-stage renal disease. Cell Death Dis. 2021 12
30. Ling HY, Song CG, Fang YW, Yin Y, Wu ZJ, Wang YH. et al. TH5487, a small molecule inhibitor of OGG1, attenuates pulmonary fibrosis by NEDD4L-mediated OGG1 degradation. Chem-Biol Interact. 2022 362
31. Gao CC, Pang LQ, Ren CC, Ma TH. Decreased expression of Nedd4L correlates with poor prognosis in gastric cancer patient. Med Oncol. 2012;29:1733-8
32. Qu MH, Han CH, Srivastava AK, Cui TT, Zou N, Gao ZQ. et al. miR-93 promotes TGF-β-induced epithelial-to-mesenchymal transition through downregulation of NEDD4L in lung cancer cells. Tumor Biol. 2016;37:5645-51
33. Wu H, Zuo JH, Dai Y, Li HR, Wang S. NEDD4 family E3 ligases in osteoporosis: mechanisms and emerging potential therapeutic targets. J Orthop Surg Res. 2025 20
34. Shen WL, Zhang ZG, Ma JQ, Lu D, Lyu L. The Ubiquitin Proteasome System and Skin Fibrosis. Mol Diagn Ther. 2021;25:29-40
35. Chen BJ, Li RS, Hernandez SC, Hanna A, Su K, Shinde AV. et al. Differential effects of Smad2 and Smad3 in regulation of macrophage phenotype and function in the infarcted myocardium. J Mol Cell Cardiol. 2022;171:1-15
36. Yan XH, Liao HW, Cheng MZ, Shi XJ, Lin X, Feng XH. et al. Smad7 Protein Interacts with Receptor-regulated Smads (R-Smads) to Inhibit Transforming Growth Factor-β (TGF-β/Smad Signaling. Journal of Biological Chemistry. 2016;291:382-92
37. Ying HJ, Fang M, Hang QQ, Chen YM, Qian X, Chen M. Pirfenidone modulates macrophage polarization and ameliorates radiation-induced lung fibrosis by inhibiting the TGF-β1/Smad3 pathway. J Cell Mol Med. 2021;25:8662-75
38. Bhattacharya M, Ramachandran P. Immunology of human fibrosis. Nat Immunol. 2023
39. Fabre T, Barron AMS, Christensen SM, Asano S, Bound K, Lech MP. et al. Identification of a broadly fibrogenic macrophage subset induced by type 3 inflammation. Sci Immunol. 2023 8
40. Deng ZQ, Fan T, Xiao C, Tian H, Zheng YJ, Li CX. et al. TGF-β signaling in health, disease, and therapeutics. Signal Transduct Tar. 2024 9
41. Chen L, Yang Y, Yan HY, Peng XY, Zou J. NEDD4L-induced β-catenin ubiquitination suppresses the formation and progression of interstitial pulmonary fibrosis inhibiting the CTHRC1/HIF-1α axis. Int J Biol Sci. 2021;17:3320-30
42. Revici R, Hosseini-Alghaderi S, Haslam F, Whiteford R, Baron M. E3 Ubiquitin Ligase Regulators of Notch Receptor Endocytosis: From Flies to Humans. Biomolecules. 2022 12
Author contact
![]() Corresponding authors: Yong He, Ph.D., School of Chinese Materia Medica, Nanjing University of Chinese Medicine or Shanghai Institute of Materia Medica (SIMM), Chinese Academy of Sciences, 555 Zuchongzhi Road, Pudong, Shanghai 201203, China; Email: heyongac.cn; Yuanwen Chen, Ph.D., MD., Department of Gastroenterology, Huadong Hospital, Fudan University, 221 West Yanan Road, Shanghai 200040, China. Email: chenywhdgiedu.cn; Peng Wang, MS., Department of Hepatobiliary Medicine, Shanghai Eastern Hepatobiliary Surgery Hospital, Naval Medical University, Shanghai 200438, China. Email: wpmoderatecom.
Corresponding authors: Yong He, Ph.D., School of Chinese Materia Medica, Nanjing University of Chinese Medicine or Shanghai Institute of Materia Medica (SIMM), Chinese Academy of Sciences, 555 Zuchongzhi Road, Pudong, Shanghai 201203, China; Email: heyongac.cn; Yuanwen Chen, Ph.D., MD., Department of Gastroenterology, Huadong Hospital, Fudan University, 221 West Yanan Road, Shanghai 200040, China. Email: chenywhdgiedu.cn; Peng Wang, MS., Department of Hepatobiliary Medicine, Shanghai Eastern Hepatobiliary Surgery Hospital, Naval Medical University, Shanghai 200438, China. Email: wpmoderatecom.