Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Hierarchical regulation of UMEs...
From cell fate modulation to...
Emerging hotspots and future...
Conclusion, limitations, and...
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(8):4194-4224. doi:10.7150/ijbs.130460 This issue Cite
Review
Ubiquitin-Modifying Enzymes as Cell-Fate Regulators in Intestinal Inflammation
Chenchen Qian1,2, Fangmin Ning1, Yong Xu1, Jingjing Shao1, Binglu Shi1, Chenjian Zhou2 ![]() , Yi Wang1
, Yi Wang1 ![]()
1. School of Pharmacy, Hangzhou Normal University, Hangzhou, Zhejiang 311121, China.
2. Pharmacy Department, Wenzhou Central Hospital, Affiliated to Wenzhou Medical University, Wenzhou, Zhejiang 325000, China.
Received 2025-12-23; Accepted 2026-3-18; Published 2026-4-8
Abstract

Inflammatory bowel disease (IBD), including ulcerative colitis (UC) and Crohn's disease (CD), is a chronic, relapsing inflammatory disorder of the gastrointestinal tract. Intestinal homeostasis relies on the intricate balance of cell fate decisions within the intestinal epithelium and immune compartments. Ubiquitin-modifying enzymes (UMEs), including E2 conjugating enzymes, E3 ubiquitin ligases, and deubiquitinating enzymes (DUBs), have emerged as pivotal molecular regulators of these processes by orchestrating post-translational modifications that dictate protein stability, activity, and localization. In this review, we systematically summarize the essential roles of UMEs in modulating diverse cell-fate outcomes and their subsequent effects on intestinal barrier integrity and immune responses. Furthermore, we discuss the pathogenic dysregulation of specific UMEs in IBD and highlight their potential as diagnostic biomarkers and therapeutic targets. Finally, we explore emerging strategies, including small-molecule inhibitors and PROTAC technology, for targeting UMEs in clinical applications. By integrating current advances, this review provides novel insights into the ubiquitin-mediated regulation of intestinal cell fate and offers new perspectives for the management of IBD and the prevention of colitis-associated cancer (CAC).
Keywords: cell fate, inflammatory bowel disease, ubiquitin-modifying enzymes, post-translational modifications
Introduction
Inflammatory bowel disease (IBD), together with other forms of chronic colitis, represents a spectrum of persistent inflammatory disorders of the gastrointestinal tract, primarily including ulcerative colitis (UC) and Crohn's disease (CD) [1, 2]. Its global incidence and prevalence have increased steadily, imposing a substantial healthcare burden [3]. The pathogenesis of chronic colitis, including IBD, involves multiple interconnected mechanisms, including disruption of the intestinal epithelial barrier, endoplasmic reticulum (ER) stress, immune dysregulation, and intestinal microbiota dysbiosis [4, 5]. Although agents targeting TNF-α and related pathways have been used in IBD treatment, their efficacy remains limited, with high recurrence rates and significant adverse effects [6]. Therefore, identifying novel molecular mechanisms and therapeutic targets remains a major priority. Moreover, emerging evidence indicates that dysregulated cell fate programs represent a key pathological convergence point linking chronic intestinal inflammation and disease progression, including IBD and colitis-associated cancer (CAC) [7].
In this disease context, “cell fate” does not refer solely to transient changes in signaling activity but also encompasses irreversible or long-lasting biological outcomes that determine tissue integrity and immune equilibrium [8]. These regulatory programs operate at both cellular and tissue levels. At the cellular level, they control epithelial cell survival or programmed cell death (including apoptosis, necroptosis, pyroptosis, and ferroptosis), intestinal stem cell renewal and differentiation, polarization and functional reprogramming of innate immune cells, and lineage commitment of adaptive immune cells [9-15]. At the tissue level, these processes collectively shape pathological outcomes, including chronic inflammation, fibrotic remodeling, dysplasia, and CAC [16, 17]. Collectively, cell fate regulation represents the terminal decision layer through which upstream inflammatory signals are translated into concrete cellular behaviors and disease-relevant pathological outcomes. Here, we emphasize that stress-adaptive responses, such as the unfolded protein response (UPR) and autophagy, are increasingly recognized as fate-modulating programs that influence cellular trajectories, rather than independent terminal fate endpoints.
Consistent with this framework, accumulating evidence indicates that ubiquitin-modifying enzymes (UMEs) act as key molecular switches linking inflammatory signaling networks to fate-determining programs. By controlling the stability, localization, and activity of core regulators involved in cell death, stress adaptation, differentiation, and immune activation, UMEs actively bias cellular trajectories toward survival versus death, repair versus injury, and immune tolerance versus chronic inflammation. Accordingly, ubiquitination and deubiquitination emerge as upstream determinants of cell fate decisions in the inflamed intestinal microenvironment.
Ubiquitin-modifying enzymes (UMEs), composed of ubiquitination enzymes (E1, E2, and E3 ligases) and deubiquitinating enzymes (DUBs), represent a central post-translational regulatory system governing protein homeostasis and signaling, thereby controlling protein fate and downstream cellular decisions [18, 19]. Emerging evidence indicates that UMEs play critical roles in IBD pathogenesis by regulating epithelial cell survival and regeneration programs, immune cell activation and differentiation, and stress adaptation responses, thereby shaping epithelial barrier homeostasis and remodeling the inflammatory microenvironment [18-20]. Several UMEs, such as A20, CYLD, OTULIN, and USP13, are aberrantly expressed in intestinal tissues from IBD patients and experimental models, and are closely associated with epithelial injury, immune dysregulation, and disease progression [21-25]. However, systematic insights into the cell-type-specific functions, context-dependent roles, and dynamic regulation of UMEs in distinct IBD subtypes remain limited.
In this review, we systematically summarize recent advances in UME-mediated regulation of cell fate during intestinal inflammation. By integrating evidence across distinct cell types and signaling contexts, we highlight how these molecular switches orchestrate the transition between survival and death, repair and injury, and immune tolerance and chronic inflammation, thereby providing a conceptual framework for therapeutic targeting.
Hierarchical regulation of UMEs in intestinal inflammation
System overview, spatial organization, and chain-type specificity
The UME system comprises four enzymatic classes: E1 (activating), E2 (conjugating), E3 (ligating), and DUBs (removing) [26]. This system enables precise regulation of protein stability, subcellular localization, interactions, and functional states, thereby coordinating diverse physiological and pathological processes [19, 27]. During the enzymatic cascade, E1 activates ubiquitin (Ub) in an ATP-dependent manner, E2 transfers ubiquitin to E3, and E3 recognizes specific substrates to catalyze ubiquitin conjugation. Conversely, DUBs remove mono- or polyubiquitin chains from target proteins, ensuring the reversibility and dynamic regulation of ubiquitin signaling (Figure 1A-B).
Roles of UMEs in IBD. (A) The ubiquitination cascade involves E1-mediated ubiquitin activation, E2-mediated transfer, and E3-catalyzed substrate modification, while DUBs remove ubiquitin and maintain signaling balance. (B) IBD susceptibility genes encode multiple UMEs, underscoring their critical roles in disease pathogenesis. (C) Distinct ubiquitin linkages (mono-, multi-mono-, and poly-ubiquitin chains such as K48, K63, and M1) confer specific cellular outcomes including degradation and signaling regulation. Created in BioRender. Qian, C. (2026) https://BioRender.com/u85g4fh.
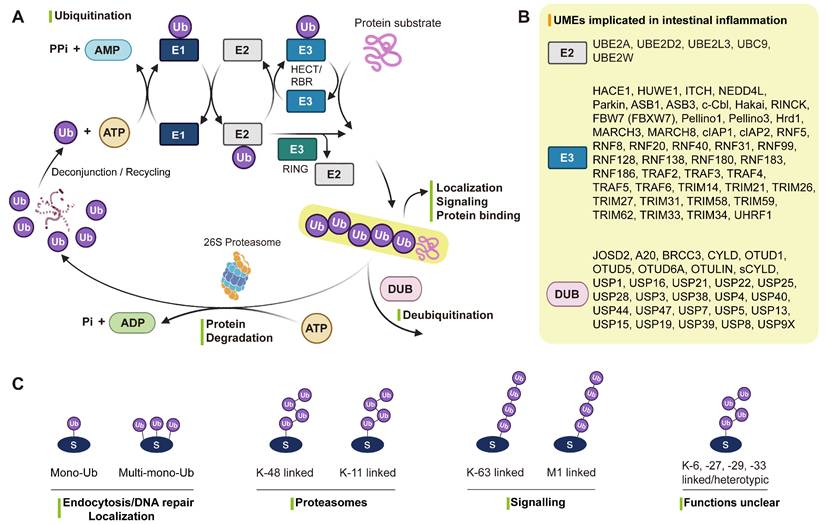
Ubiquitin can be linked through seven lysine residues (K6, K11, K27, K29, K33, K48, K63) or the N-terminal methionine (M1) to form distinct chain types [28] (Figure 1C). Among them, K48/K11-linked chains mainly mediate proteasomal degradation [29, 30], while K63/M1-linked chains regulate inflammation and immune signaling [31]. In contrast, mono-ubiquitination and atypical linkages such as K6, K27, K29, and K33 are involved in endocytosis, DNA repair, and vesicular trafficking [32]. In IBD, the balance between different chain types and their removal by UMEs is finely tuned to control immune signaling, cell death, autophagy, and epithelial barrier integrity [33]. Disruption of this dynamic equilibrium represents a central molecular mechanism driving chronic intestinal inflammation [33].
The intestinal immune system primarily comprises intestinal epithelial cells, innate immune cells, and adaptive immune cells (Figure 2A). Within this organized cellular landscape, UMEs display distinct cell type-specific distributions and functional roles (Figure 2B-D), forming a multilayered regulatory architecture that coordinates epithelial barrier homeostasis and immune responses. This spatial and functional stratification provides a structural basis for the fine-tuned regulation of ubiquitin signaling during intestinal inflammation.
Cell fate-layered landscape of UME functions in intestinal inflammation. (A) Schematic overview of major cellular compartments involved in intestinal inflammation, including intestinal epithelial cells, innate immune cells, and adaptive immune cells across the epithelial barrier and lamina propria. (B) Summary of reported functions of E2 ubiquitin-conjugating enzymes across epithelial, innate immune, and adaptive immune compartments. (C) Classification of E3 ubiquitin ligases according to their reported protective or pathogenic roles in regulating epithelial and immune cell fate decisions. (D) Classification of DUBs based on their functional impact on inflammatory and stress-responsive pathways across distinct cellular compartments. Protective roles refer to functions that promote epithelial barrier integrity, immune homeostasis, resolution of inflammation, or tumor-suppressive outcomes, whereas pathogenic roles indicate activities that amplify inflammatory signaling, impair barrier function, or facilitate chronic inflammation and colitis-associated tumorigenesis. UMEs marked with asterisks (*) exhibit context-dependent or bidirectional effects depending on cell type, inflammatory stimulus, or disease stage. “Not yet studied” indicates the absence of direct experimental evidence in the corresponding cellular compartment rather than confirmed lack of function. Created in BioRender. Qian, C. (2026) https://BioRender.com/9xtir6c.
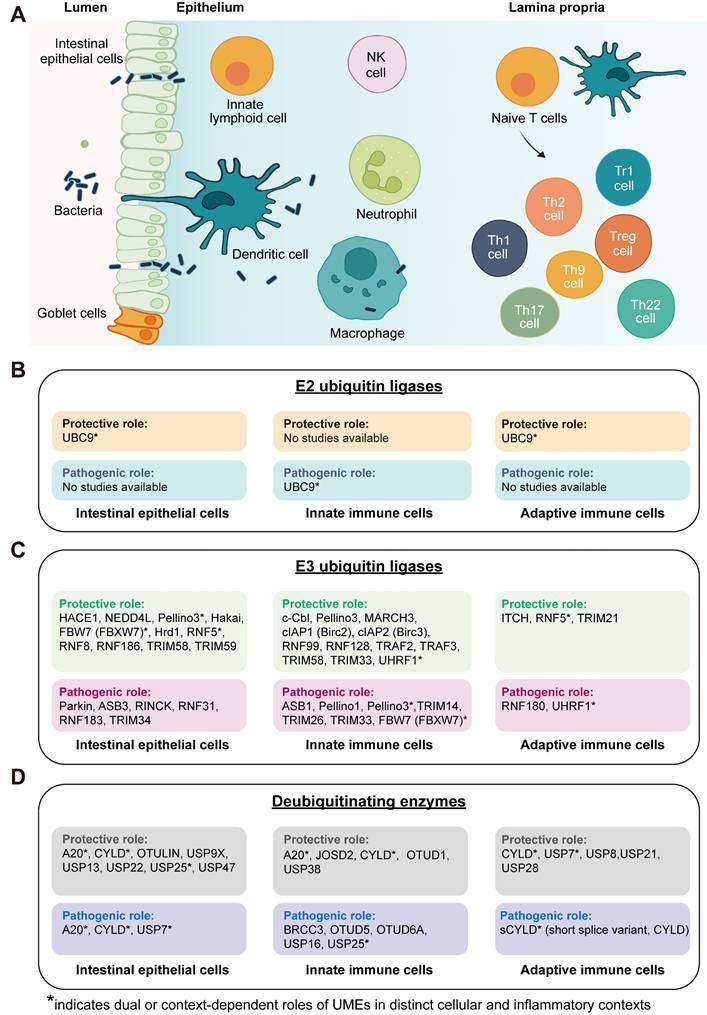
Hierarchical regulation of cell fate by E2 ubiquitin-conjugating enzymes
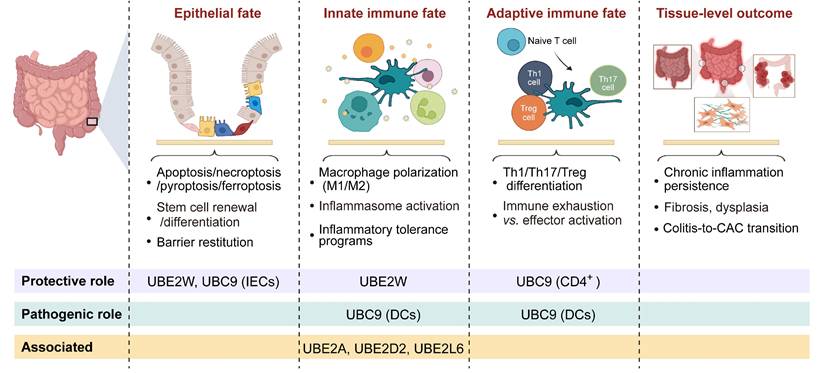
E2 ubiquitin-conjugating enzymes constitute the core module of the ubiquitination cascade, determining ubiquitin chain elongation modes, linkage specificity, and substrate fate [34]. Beyond their enzymatic activity, accumulating evidence indicates that E2 enzymes also modulate substrate stability and signaling complex assembly, thereby regulating the amplitude and duration of inflammatory and stress-responsive pathways [35-38]. In intestinal inflammation, E2-mediated regulation primarily tunes the amplitude of inflammatory signaling and the cellular activation thresholds, thereby influencing epithelial stress tolerance, antigen presentation, and immune polarization. Within the hierarchical cellular-to-tissue regulatory framework defined above (Tables 1-2), E2 functions can be systematically mapped onto epithelial, innate immune, adaptive immune, and tissue outcome layers, highlighting their coordinated contributions to inflammatory progression and tissue remodeling (Figure 2B, Figure 3, Table 3).
Operational definition of cell-fate programs in intestinal inflammation.
| Cell fate layer | Operational definition | Representative processes | Pathological relevance | Reference |
|---|---|---|---|---|
| Epithelial fate | Determines epithelial survival, elimination, and regenerative capacity under inflammatory stress | Apoptosis, necroptosis, pyroptosis, ferroptosis, intestinal stem cell renewal and differentiation, barrier restitution | Barrier disruption, impaired mucosal repair, accumulation of genetically damaged epithelial cells | [9, 10] |
| Innate immune fate | Defines functional states and inflammatory thresholds of innate immune cells | Macrophage polarization (M1/M2), inflammasome activation, inflammatory tolerance programs | Amplification or resolution of chronic inflammation, shaping tumor-promoting inflammatory microenvironment | [11-13] |
| Adaptive immune fate | Controls lineage commitment and effector status of adaptive immune cells | Th1/Th17/Treg differentiation, immune exhaustion versus effector activation | Immune imbalance, impaired immune surveillance, sustained pro-tumorigenic inflammation | [14, 15] |
Tissue-level inflammatory and carcinogenic outcomes emerging from cumulative cell-fate programs.
| Tissue-level outcome state | Integrated cellular drivers | Representative tissue features | Disease relevance | Reference |
|---|---|---|---|---|
| Inflammatory resolution and mucosal restoration | Balanced epithelial regeneration + restrained innate immune activation + regulatory adaptive immunity | Barrier repair, reduced inflammatory infiltrates, restoration of tissue architecture | Disease remission, reduced relapse risk | [16] |
| Chronic inflammatory remodeling | Persistent epithelial injury + pro-inflammatory innate polarization + immune imbalance | Sustained inflammation, fibrosis, crypt architectural distortion, dysplasia-prone microenvironment | Disease progression, increased CAC susceptibility | [17] |
| Tumor-permissive tissue state (CAC-prone) | Aberrant epithelial survival + genomic instability + pro-tumor immune niche formation | Dysplasia, epithelial transformation, stromal remodeling, pro-angiogenic signaling | CAC initiation and malignant progression | [17] |
Cell fate-layered organization of E2 ubiquitin-conjugating enzyme functions in intestinal inflammation. Schematic illustrates the hierarchical organization of E2 enzyme-mediated regulation across interconnected cellular fate layers and downstream tissue-level outcomes during intestinal inflammation. Cellular fate layers include epithelial fate, innate immune fate, and adaptive immune fate, whereas tissue-level outcomes represent the integrated pathological consequences arising from the cumulative effects of these cellular programs. Representative biological processes regulated at each layer are indicated, including epithelial cell death and barrier restitution, macrophage polarization and inflammasome activation, T cell differentiation and immune exhaustion, as well as tissue-scale outcomes such as chronic inflammation and colitis-associated cancer (CAC) progression. Created in BioRender. Qian, C. (2026) https://BioRender.com/d0r43h0.
Roles of E2 ubiquitin-conjugating enzymes in intestinal inflammation and tumorigenesis.
| Gene | Pathogenesis of IBD/CAC | Effect | Cell fate | Mechanism/major finding | Target protein classification | Cell type | Alteration in patients | Transgenic mice | Disease model | Disease Phenotype | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UBE2W | Imbalance of intestinal immunity | Anti-inflammatory | Innate immune fate (inflammatory activation threshold); Epithelial fate (barrier injury susceptibility) | UBE2W limits NF-κB activation by reducing IκB and p65 phosphorylation and preventing p65 nuclear translocation, thereby lowering pro-inflammatory cytokine expression | - | - | - | AAV2/9-Ube2w OE | DSS | Attenuated colitis | [40] |
| UBE2L3/ UBCH7 | Genetic susceptibility | - | - | - | - | - | - | - | - | - | [165] |
| UBE2A, UBE2D2, UBE2L6 | Immune tolerance and inflammatory activation | - | Innate immune fate (macrophage activation versus tolerogenic state) | - | - | - | - | - | - | - | [39] |
| UBC9 (SUMO E2) | Imbalance of intestinal immunity | Anti-inflammatory; Th17 pathogenicity-restraining | Adaptive immune fate (Th17 effector programming) | Hypoxia-induced HIF-1α binds to the Ubc9 promoter CpG island and promotes DNA hypermethylation, leading to transcriptional repression of Ubc9, reduced SUMOylation of RORγt, enhanced IL-17 transcriptional activity, and reinforcement of pathogenic Th17 effector responses via hypoxia-driven epigenetic reprogramming. | RORγt (SUMOylation substrate) | CD4⁺ Th17 cells; colonic lamina propria lymphocytes | Down-regulated in UC patients | - | - | - | [41] |
| UBC9 (SUMO E2) | Defect of intestinal barrier, imbalance of intestinal immunity | Anti-inflammatory; epithelial barrier-protective | Epithelial fate (survival and barrier maintenance) | Ubc9 downregulation reduces Akt1 SUMOylation and stability, leading to enhanced NF-κB-dependent inflammatory gene expression and impaired wound-healing responses through SUMO-mediated regulation of cellular stress tolerance. | Akt1 (SUMOylation substrate) | IECs, HCT-8 | Down-regulated in UC and CD biopsies | Not genetic KO; Ubc9HyperLow | DSS | Susceptibility to colitis | [42] |
| UBC9 (SUMO E2) | Imbalance of intestinal immunity | Pro-inflammatory | Innate immune fate (DC activation state); Adaptive immune fate (CD4⁺ T cell priming and polarization) | Ubc9-dependent SUMOylation of RBPJ stabilizes RBPJ by blocking proteasomal degradation, leading to enhanced Ciita-driven MHC class II transcription and increased antigen presentation capacity of DCs, which promotes CD4⁺ T cell activation and Th1/Th17 polarization | RBPJ (SUMOylation substrate) | DCs (BMDCs, CD11c⁺ DCs) | - | Itgax-Cre; Ubc9f/f (DC-specific KO) | DSS | Attenuated colitis | [43] |
In intestinal inflammation, available evidence suggests that E2-mediated regulation primarily modulates the intensity of inflammatory signaling and the cellular activation thresholds. Gene expression analyses revealed that E2 enzymes, including UBE2A, UBE2D2, and UBE2L6, are differentially expressed in intestinal macrophages under inflammatory conditions in the gut [39]. UBE2W attenuates DSS-induced colitis by limiting NF-κB-driven inflammatory amplification and epithelial barrier damage [40]. UBC9 exhibits cell type-specific effects: in CD4⁺ T cells, reduced UBC9 enhances pathogenic Th17 responses [41]; in IECs, UBC9 downregulation exacerbates NF-κB-dependent inflammation and colitis susceptibility [42]; whereas in DCs, UBC9-mediated SUMOylation of RBPJ promotes T-cell activation and Th1/Th17 polarization, with DC-specific Ubc9 deletion alleviating experimental colitis [43]. Collectively, these findings suggest that E2 enzymes act as critical regulators of intestinal immune homeostasis, influencing epithelial barrier integrity and immune cell polarization (Figure 3, Table 3). Notably, compared with the extensive characterization of E3 ligases and DUBs, current knowledge of E2 enzymes in intestinal inflammation remains limited and largely confined to a small subset of family members. Thus, E2-mediated regulation should be viewed as an emerging regulatory layer, with substantial gaps remaining in mechanistic and disease-level understanding.
Hierarchical regulation of cell fate by E3 ligases
E3 ubiquitin ligases function as molecular switches that govern the stability and activity of key regulatory proteins during inflammation.
Unlike E2 enzymes that primarily tune activation thresholds, E3 ligases control signal directionality and strength, thereby shaping distinct cellular programs. In intestinal inflammation, they coordinate stress responses, survival, and cell death across multiple signaling axes, forming a multilayered regulatory framework that links molecular events to tissue-level outcomes (Figure 2C, Figure 4A, Table 4). Structurally, the E3 ligases discussed encompass major classes, including RING, HECT, RBR, and non-classical types [44].
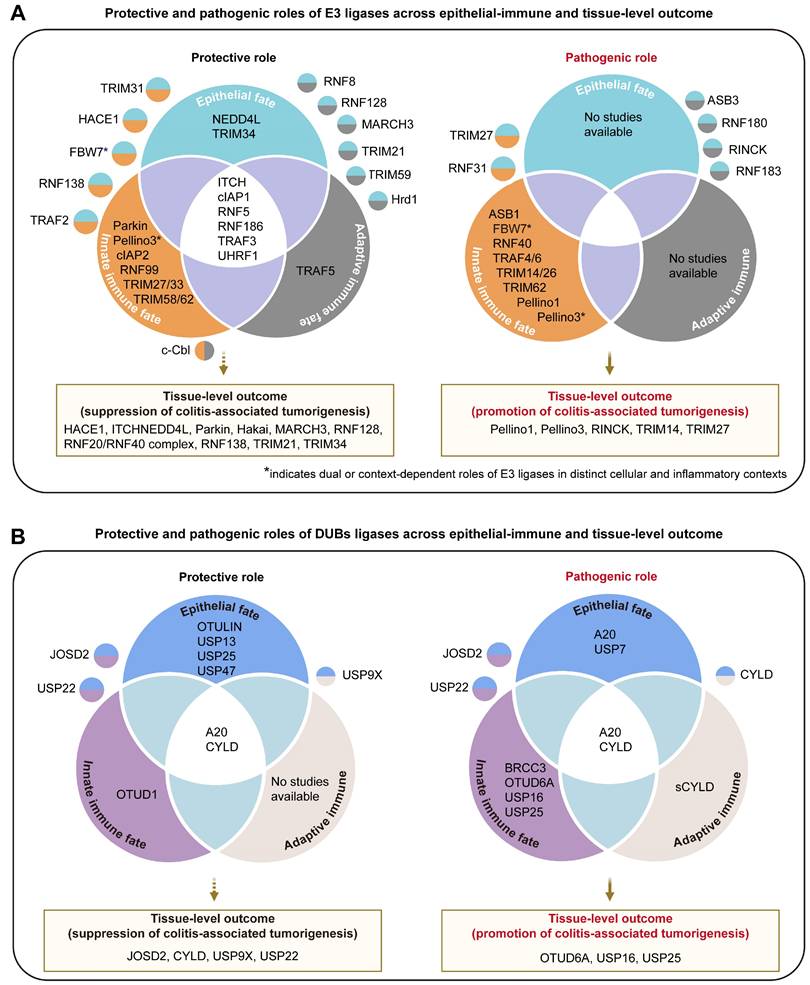
Protective and pathogenic roles of UMEs in epithelial-immune cell fate layers and tissue-level outcomes during intestinal inflammation. (A) Summary of E3 ligases and (B) deubiquitinases (DUBs) categorized according to reported protective or pathogenic functions across distinct cell fate layers, including epithelial, innate immune, and adaptive immune fate layers, together with their associated tissue-level outcomes related to colitis-associated cancer (CAC) progression. Venn diagrams illustrate UMEs that act within single compartments or coordinately regulate multiple cellular layers. UMEs shown in overlapping regions indicate shared regulatory roles across epithelial and immune compartments. Tissue-level fate panels summarize enzymes implicated in promoting malignant transition or suppressing inflammation-driven tumorigenesis. UMEs marked with asterisks (*) exhibit context-dependent or bidirectional effects depending on cell type, inflammatory stimulus, or disease stage.
Roles of E3 ubiquitin ligases in intestinal inflammation and tumorigenesis.
| E3 Type | Gene | Pathogenesis of IBD/CAC | Effect | Cell fate | Mechanism/major finding | Target protein classification | Cell type | Alteration in patients | Transgenic mice | Disease model | Disease Phenotype | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HECT | HACE1 | Imbalance of intestinal immunity | Anti-inflammatory; CAC initiation-suppressive | Epithelial fate (apoptosis, necroptosis); Innate immune fate (NF-κB) | HACE1 ubiquitinates TRAF2 to modulate TNFR1 complex signaling, thereby limiting TNFα-induced systemic inflammation | TRAF2 (ubiquitination substrate, K63-linked) | IECs | - | Hace1-/- | DSS; AOM/DSS | Susceptibility to colitis and CAC | [46] |
| HECT | HUWE1 | - | - | Epithelial fate (goblet cell differentiation) | HUWE1 negatively regulates goblet cell generation by promoting ATOH1 degradation | - | Goblet cells | - | - | - | - | [166] |
| HECT | ITCH | Imbalance of intestinal immunity | Anti-inflammatory | Innate immune fate; Adaptive immune fate (Th17 effector programming) | ITCH deficiency leads to dysregulated gut microbiota and exaggerated T cell activation, resulting in spontaneous colitis | RORγt (ubiquitination substrate) | CD4⁺ T cells; intestinal immune cells | - | Itch-/- | DSS | Severe colitis | [55] |
| HECT | ITCH | Defect of intestinal barrier; imbalance of intestinal immunity | Anti-inflammatory; CAC initiation-suppressive | Adaptive immune fate (Th17 differentiation); Epithelial fate (barrier restitution) | ITCH ubiquitinates and degrades RORγt, suppressing Th17 differentiation and IL-17-mediated intestinal inflammation and tumorigenesis. ITCH deficiency leads to exaggerated Th17 responses and enhanced NF-κB signaling in the gut | RORγt (ubiquitination substrate, K48-linked) | CD4⁺ T cells (Th17 cells) | - | Itch-/- | DSS; AOM/DSS | Susceptibility to colitis and CAC | [54] |
| HECT | ITCH | Imbalance of intestinal immunity | Anti-fibrotic | Innate immune fate; Adaptive immune fate (Th17 differentiation) | ITCH ubiquitinates and degrades HIC-5, suppressing IL-17-driven fibroblast activation and extracellular matrix production. ITCH deficiency results in increased HIC-5 stability and exacerbated intestinal fibrosis | HIC-5 (ubiquitination substrate, K63-linked) | Intestinal fibroblasts | - | Itch-/- | DSS-induced chronic colitis/fibrosis model | Severe intestinal fibrosis | [90] |
| HECT | NEDD4L | Defect of intestinal barrier; imbalance of intestinal immunity | Anti-inflammatory; barrier-protective; CAC initiation-suppressive | Epithelial fate (ferroptosis, epithelial death, barrier restitution) | NEDD4L promotes ferroptosis in intestinal epithelial cells by maintaining the SLC3A2-GPX4 axis suppression, thereby limiting IEC proliferation, resolving inflammation, and inhibiting colorectal tumorigenesis | SLC3A2 (ubiquitination substrate, K63-linked) | IECs; intestinal organoids | Down-regulated in intestinal mucosa | Nedd4l+/-; Nedd4lfl/fl VillinCre | DSS; TNBS; AOM/DSS | Severe colitis; prone to CAC | [47] |
| RBR | Parkin | Imbalance of intestinal immunity | Pro-inflammatory; barrier-disruptive | Epithelial fate (barrier maintenance impairment) | Parkin promotes autophagy-lysosome-mediated degradation of VDR, leading to reduced VDR signaling, impaired epithelial barrier integrity | VDR (ubiquitination substrate) | IECs | - | Parkin-/- | DSS | Attenuated colitis | [64] |
| RBR | Parkin | Imbalance of intestinal immunity | CAC initiation-suppressive | Innate immune fate | Parkin mediates ubiquitination and degradation of ITF2; NF-κB p65 competes with Parkin to stabilize ITF2, thereby suppressing NF-κB target genes and inhibiting CAC progression | ITF2 (ubiquitination substrate, K48-linked) | IECs | - | - | - | - | [167] |
| RING-like (non-classical) | ASB1 | Imbalance of intestinal immunity | Pro-inflammatory | Innate immune fate | ASB1 binds and stabilizes TAB2, thereby enhancing TAK1-dependent activation of NF-κB and MAPK signaling and promoting pro-inflammatory cytokine production | TAB2 (signaling adaptor/binding partner) | BMDMs; BMDCs | - | Asb1-/- | DSS | Attenuated colitis | [127] |
| RING-like (non-classical) | ASB3 | Disturbance of gut microbiota; imbalance of intestinal immunity | Pro-inflammatory | Epithelial fate; Innate/adaptive immune fate | ASB3 promotes the ubiquitination and degradation of TRAF6 in intestinal epithelial cells, leading to aberrant NF-κB activation and intestinal microbiota imbalance | TRAF6 (ubiquitination substrate, K48-linked) | IECs | Up-regulated in IBD patients | ASB3-/-; ASB3OE (IEC) | DSS | Attenuated colitis (ASB3-/-); aggravated colitis (ASB3OE (IEC)) | [123] |
| RING-like (non-classical) | c-Cbl | Imbalance of intestinal immunity; impaired tolerogenic DC function | Anti-inflammatory (restricts DSS colitis) | Innate/adaptive immune fate | c-Cbl mediates ubiquitination and degradation of RelB downstream of Dectin-2/3 in DCs; c-Cbl deficiency leads to RelB activation, which suppresses IL-10 transcription | RelB (ubiquitination substrate) | DCs, macrophages | Down-regulated in intestinal mucosa | c-Cblf/fCD11cCre/+ | DSS; Gut fungi manipulation | Severe DSS colitis; fungi-dependent inflammation amplification | [78] |
| SCF-type Cullin-RING E3 | FBW7 (FBXW7) | Defect of intestinal barrier; imbalance of intestinal immunity | Anti-inflammatory | Innate immune fate (epithelial NF-κB hyperactivation) | IEC-specific FBW7 deletion activates NF-κB pathway (↑TNFα, IL-6, IL-1β), aggravated epithelial damage, and exacerbated colitis severity | - | IECs | - | Fbw7ΔG (Vil/Cre; Fbw7fl/fl) | DSS | Severe colitis | [168] |
| SCF-type Cullin-RING E3 | FBW7 (FBXW7) | Defect of intestinal barrier; imbalance of intestinal immunity | Pro-inflammatory | Innate immune fate | FBW7 directly ubiquitinates EZH2, reducing CCL2/7 expression and limiting CX3CR1hit macrophage recruitment | EZH2 (ubiquitination substrate, K48-linked) | Macrophages | Up-regulated in intestinal mucosa | LysM+Fbxw7fl/fl; AAV-shFbxw7 | DSS; TNBS | Attenuated colitis (LysM+Fbxw7fl/fl); aggravated colitis (AAV-shFbxw7) | [169] |
| RING-like (non-classical) | Pellino1 | Imbalance of intestinal immunity | Pro-inflammatory; CAC initiation-promoting | Innate immune fate (macrophage activation amplification, M2-like polarization bias) | Pellino1 ubiquitinates and stabilizes STAT3 in intestinal macrophages, enhancing STAT3 activation and amplifying macrophage-mediated inflammatory signaling to promote a pathogenic intestinal environment | STAT3 (ubiquitination substrate, K63-linked) | Macrophages | Up-regulated in colonic mucosa | Pellino1-mKO | DSS; AOM/DSS | Attenuated colitis; reduced CAC | [52] |
| RING-like (non-classical) | Pellino3 | Imbalance of intestinal immunity | Pro-inflammatory; CAC initiation-promoting | Innate immune fate (TLR4-NF-κB/MAPK) | Pellino3 inhibits IRF4-mediated negative regulation of TLR4 signaling, thereby enhancing TLR4-driven inflammation. Loss of Pellino3 reduces colitis severity, lowers inflammation-induced colorectal tumor burden, and decreases activation of NF-κB, STAT3, and ERK pathways | IRF4 (signaling adaptor/binding partner) | Macrophages | - | Peli3-/- | DSS; AOM/DSS | Attenuated colitis; reduced CAC | [53] |
| RING-like (non-classical) | Pellino3 | Imbalance of intestinal immunity | Anti-inflammatory | Innate immune fate (NF-κB/MAPK) | Pellino directly binds RIP2 via the FHA domain and catalyzes K63-linked ubiquitination through its RING-like domain; promotes TAK1/IKK recruitment, NF-κB/MAPK activation, enabling Nod2 protective signaling | RIP2 (ubiquitination substrate, K63-linked) | IECs; macrophages | Down-regulated in intestinal mucosa of CD patients | Peli3-/- | DSS; TNBS; C. rodentium infection | Severe colitis | [122] |
| RING-like (non-classical) | RINCK | Imbalance of intestinal immunity | Pro-inflammatory; CAC initiation-promoting | Epithelial fate (epithelial survival, barrier restitution) | IEC-specific deletion of Rinck markedly suppresses ROS production, oxidative stress, and inflammation, while overexpression of Rinck in IECs significantly exacerbates OTA/DSS-induced acute and chronic colitis | NRF2 (ubiquitination substrate, K48-linked) | IECs | Up-regulated in IBD and CRC patients | IEC-Rinck (KO); IEC-Rinck (OE) | OTA/DSS; AOM/DSS | Attenuated colitis (KO); exacerbated colitis (OE) | [68] |
| RING-like (non-classical) | Hakai | - | Anti-inflammatory; CAC initiation-suppressive | - | Hakai regulates FASN ubiquitination and lysosomal degradation, thereby limiting FASN-mediated lipid accumulation and linking lipid metabolism to IBD/CAC pathogenesis | FASN (ubiquitination substrate) | IECs | Up-regulated in colonic mucosa of UC and CD patients | - | DSS; AOM/DSS; IL-10-KO spontaneous colitis | - | [70] |
| RING | MARCH3 | Imbalance of intestinal immunity | Anti-inflammatory; CAC initiation-suppressive | Epithelial fate (barrier homeostasis maintenance) | MARCH3 ubiquitinates IL-6Rα (K401) and gp130 (K849) to drive receptor internalization and lysosomal degradation, thereby suppressing IL-6/OSM-STAT3 pro-inflammatory signaling and limiting colitis and CAC progression | IL-6Rα (ubiquitination substrate, K48- and K63-linked); gp130 (ubiquitination substrate, K48-linked) | Macrophages | Down-regulated in CRC tissues | March3-/- | DSS; AOM/DSS | Severe colitis; aggravated CAC | [72] |
| RING | MARCH8 | Imbalance of intestinal immunity | Inflammasome-suppressive | Innate immune fate | VANGL2 recruits MARCH8 to catalyze K27-linked poly-Ub of NLRP3 (K823), driving OPTN-mediated selective autophagic degradation and inhibiting NLRP3 inflammasome activation | NLRP3 (ubiquitination substrate, K27-linked) | - | - | - | - | - | [65] |
| RING | cIAP1 (Birc2), cIAP2 (Birc3) | Imbalance of intestinal immunity | Anti-inflammatory (Birc3) | Innate immune fate (NF-κB/MAPK) | cIAP1/2 act as RING E3 ligases for RIP2, mediating its K63-linked polyubiquitination and recruitment of TAK1/TAB complexes, thereby activating NOD1/2-induced MAPK (JNK, p38) and NF-κB signaling to drive cytokine and chemokine production | RIP2 (ubiquitination substrate, K63-linked) | BMDMs; HT29 | - | Birc2-/-; Birc3-/- | DSS colitis + systemic MDP administration | Severe colitis (Birc3-/-) | [83] |
| RING | cIAP1 (Birc2) | Defect of intestinal barrier | Anti-apoptotic | Epithelial fate (TNF-induced apoptosis); Stress-adaptive fate | cIAP1 restrains TNF-induced IEC apoptosis; loss or destabilization of cIAP1 (via TWEAK signaling or Smac-mimetics) sensitizes IECs to TNF-mediated cell death and exacerbates TNF-driven enteropathies, whereas inhibition of TWEAK improves colitis | - | YAMC; MC38; macrophage | - | Birc2-/-; Birc2-/-Tnfrsf1a-/- | TNF-induced enteropathy model | Susceptibility to TNF-induced cell death | [61] |
| RING | Hrd1 | Defect of intestinal barrier; imbalance of intestinal immunity | Anti-inflammatory; anti-apoptotic | Epithelial fate (epithelial survival and barrier maintenance) | Hrd1 activation decreases GRP78, PERK, CHOP, caspase-12, thereby limiting ER stress-induced epithelial apoptosis and inflammation | - | IECs | Down-regulated in inflamed intestinal epithelium of IBD patients | - | DSS; TNBS | Severe colitis (Hrd1 inhibitor LS102 treatment) | [62] |
| RING | RNF5 | Imbalance of intestinal immunity | Anti-inflammatory; limits epithelial-derived DAMP amplification | Epithelial fate (apoptosis); Innate immune fate (DC activation threshold, NF-κB); Adaptive immune fate (Th1) | RNF5 mediates ubiquitination and proteasomal degradation of S100A8 in IECs; RNF5 deficiency leads to increased S100A8 secretion, induction of mucosal CD4⁺ T cells, Th1-mediated pro-inflammatory responses. | S100A8 (ubiquitination substrate) | IECs; dendritic cells; CD4+ T cells | Down-regulated in intestinal mucosa of IBD patients | Rnf5-/- | DSS | Severe colitis | [56] |
| RING | RNF8 | Defect of intestinal barrier; imbalance of intestinal immunity | Anti-inflammatory | Epithelial fate (autophagy-dependent barrier maintenance) | RNF8 directly binds to AKT1 and mediates its ubiquitination, thereby suppressing AKT/mTOR signaling and restoring autophagy, and reducing pro-inflammatory cytokines | AKT1 (ubiquitination substrate) | IECs (colon epithelium); HT-29 | Down-regulated in colon tissues from UC patients (GSE36807) | LV-RNF8 (RNF8 OE) | TNBS | Attenuated colitis | [66] |
| RING | RNF20 | Imbalance of intestinal immunity | Anti-inflammatory; CAC initiation-suppressive | Epigenetic stability; inflammatory transcription control fate | RNF20 maintains H2Bub1 to restrain NF-κB activation; RNF20 loss reduces H2Bub1, increases p65 recruitment, decreases H3K9me3, elevates pro-inflammatory cytokine transcription | H2Bub1 | MCF10A | Down-regulated in colonic mucosa of UC and CAC patients | Rnf20+/- | DSS; AOM/DSS | Susceptibility to colitis and CAC | [128] |
| RING | RNF40 | Imbalance of intestinal immunity | Pro-inflammatory; CAC initiation-promoting (in intestinal epithelium) | Innate immune fate (NF-κB activation threshold) | Intestinal epithelial RNF40 sustains NF-κB signaling and promotes tumor-associated gene expression; epithelial RNF40 deletion suppresses NF-κB activity, attenuates DSS-induced colitis, and reduces tumorigenic potential | - | Colorectal cancer cell lines | - | CAC-Cre; Rnf40flox | DSS | Attenuated colitis | [80] |
| RING | RNF20/ RNF40 complex | Imbalance of intestinal immunity | Anti-inflammatory (mainly RNF20-dependent); CAC initiation-suppressive | - | The RNF20/RNF40 complex maintains H2Bub1; reduced complex activity decreases H2Bub1 and increases susceptibility to inflammation and CAC | - | - | RNF20 (but not RNF40) down-regulated in UC and CRC patients | - | DSS | Severe colitis | [80, 128] |
| RING | RNF31 | Imbalance of intestinal immunity | Pro-inflammatory; epithelial dysfunction | Epithelial fate (barrier integrity and epithelial dysfunction) | RNF31 knockdown stabilizes NRF2, thereby enhancing oxidative stress defense, whereas NRF2 loss impairs epithelial barrier integrity and aggravates mucosal inflammation | NRF2 (ubiquitination substrate, K63-linked) | IECs | Up-regulated in UC patients | RNF31-knockdown | DSS | Attenuated colitis | [69] |
| RING | RNF31 | Defect of intestinal barrier; imbalance of intestinal immunity | Pro-inflammatory | Innate immune fate (NLRP3 inflammasome activation); Epigenetic stability (tight junction loss) | RNF31 promotes NLRP3 inflammasome activation through ubiquitin-dependent regulation, thereby amplifying innate inflammatory responses and exacerbating intestinal inflammation | NLRP3 (ubiquitination substrate, K63-linked) | - | Up-regulated in UC patients | Adenovirus-mediated RNF31 knockdown | DSS | Attenuated colitis | [76] |
| RING | RNF99 | Imbalance of intestinal immunity | Anti-inflammatory | Innate immune fate (macrophage inflammatory activation control) | RNF99 catalyzes ubiquitination of TAB2 at K611, promotes TAB2 proteasomal degradation, suppresses TAK1-NF-κB/MAPK signaling, and reduces pro-inflammatory cytokine production | TAB2 (ubiquitination substrate, K48-linked) | PMs, BMDMs | Down-regulated in CD14⁺ monocytes from Gram-negative-infected patients | RNF99-/- | DSS | Severe colitis | [84] |
| RING | RNF128 | Imbalance of intestinal immunity | Anti-inflammatory | Immune homeostasis fate | RNF128 binds S100A8 and promotes its K63-linked ubiquitination at K36, which allows the autophagy receptor Tollip to recognize S100A8 and target it for selective autophagic degradation. Loss of RNF128 suppresses S100A8-Tollip-mediated autophagy, leading to S100A8 accumulation, enhanced macrophage cytokine production | S100A8 (ubiquitination substrate, K63-linked) | BMDMs, THP-1 | Down-regulated in inflamed colonic macrophages (IBD) | Rnf128-/- | DSS; TNBS | Severe colitis | [67] |
| RING | RNF128 | Imbalance of intestinal immunity; IL-6-STAT3 hyperactivation | Anti-inflammatory; CAC initiation-suppressive | Epithelial fate (proliferation restraint) | RNF128 ubiquitinates IL-6Rα and gp130 (K48-linked), promotes lysosomal degradation, suppresses IL-6-STAT3 signaling, thereby limiting epithelial hyperproliferation, inflammatory amplification and colitis-to-CAC transition | IL-6Rα and gp130 (ubiquitination substrate, K48-linked) | BMDMs, HCT116, SW620 | Down-regulated in colitis and CRC tissues | RNF128-/-; Apcmin/+/Rnf128+/- | DSS; AOM/DSS; APCmin/+ | Severe colitis; aggravated CAC | [89] |
| RING | RNF138 | Colitis-to-tumor transition dysregulation | Anti-inflammatory; CAC initiation-suppressive | Innate immune fate (NF-κB); Epithelial fate (epithelial proliferation restraint) | RNF138 suppresses NF-κB activation by binding NIBP (NIK/IKKβ-binding protein) and retaining NIBP in the nucleus, thereby preventing NIBP-IKKβ cytoplasmic association and limiting p65 phosphorylation and nuclear translocation | - | HCT116, RKO | Down-regulated in CRC tissues | RNF138-/- | DSS; AOM/DSS | Severe colitis; enhanced CAC progression | [85] |
| RING | RNF180 | Imbalance of intestinal immunity | Pro-inflammatory | Adaptive immune fate (Th17/Treg skewing); Epithelial fate (barrier dysfunction) | RNF180 ubiquitinates and downregulates ALKBH5, reducing ALKBH5-mediated m6A suppression on SMARCA5, leading to SMARCA5 upregulation. The increased SMARCA5 aggravates colon inflammation and induces Th17/Treg imbalance. RNF180 knockdown reverses this axis and ameliorates UC | ALKBH5 (ubiquitination substrate) | Splenic CD4⁺ T; MLN CD4⁺ T | - | sh-RNF180 | DSS | Attenuated colitis | [170] |
| RING | RNF183 | Epithelial stress response; mucosal barrier dysfunction | Pro-inflammatory; pro-apoptotic | Stress-adaptive fate (early epithelial stress response); Apoptotic fate (TRAIL-caspase axis) | RNF183, specifically induced in intestinal epithelial cells during early colitis, binds DR5 and mediates its K63-linked ubiquitination, promoting DR5 lysosomal trafficking and enhancing TRAIL-induced caspase-8/3 activation and epithelial apoptosis | DR5 (ubiquitination substrate, K63-linked) | IECs | Up-regulated in UC and CD inflamed colon tissues | - | DSS | - | [63] |
| RING | RNF183 | Imbalance of intestinal immunity | Pro-inflammatory | Epithelial fate (barrier disruption) | RNF183 promotes intestinal inflammation by ubiquitinating and degrading IκBα, thereby activating the NF-κB pathway. RNF183 expression is negatively regulated by miR-7, which is down-regulated in IBD, leading to increased RNF183 and enhanced NF-κB-driven inflammatory responses | IκBα (ubiquitination substrate) | IECs | Up-regulated in UC and CD inflamed colon tissues | - | TNBS | - | [147] |
| RING | RNF186 | ER stress-associated epithelial injury | Pro-inflammatory; pro-apoptotic | - | RNF186 localizes to ER and ubiquitinates BNip1 (K29/K63-linked), promoting BNip1 mitochondrial translocation, ER Ca²⁺ release, UPR activation (BiP, CHOP), caspase-12/-9 activation and ER stress-mediated apoptosis | BNip1 (ubiquitination substrate, K29/K63-linked) | - | - | - | - | - | [91] |
| RING | RNF186 | Imbalance of intestinal immunity | Anti-inflammatory | Epithelial fate (autophagy maintenance) | RNF186 ubiquitinates EPHB2 (K892), enabling EFNB1-triggered, ULK1/PtdIns3K/ATG5-dependent autophagy in colonic epithelial cells, which promotes bacterial clearance and maintains mucosal homeostasis; loss of RNF186 impairs EPHB2-mediated autophagy | EPHB2 (ubiquitination substrate, K27-linked); EPHB3 (ubiquitination substrate, K48/K63-linked) | IECs, Ls174t, Caco2 | Down-regulated in UC patients | Rnf186-/- | DSS | Susceptibility to colitis | [49] |
| RING | RNF186 | Genetic susceptibility; imbalance of intestinal immunity; microbial clearance | Anti-inflammatory | Innate immune functional fate (antimicrobial response) | RNF186 ubiquitinates ATF6 at K152 to activate ATF6-UPR signaling, strengthening innate receptor responses and protecting the intestine from DSS-induced injury. RNF186 deficiency or IBD-risk variants impair ATF6 ubiquitination, reduce UPR signaling and antimicrobial immunity, leading to aggravated intestinal injury | ATF6 (ubiquitination substrate) | MDMs, BMDM, LPMs | - | RNF186 deficient (siRNA knockdown) | DSS | Severe colitis | [92] |
| RING | RNF186 | Intestinal barrier dysfunction; ER stress dysregulation | Anti-inflammatory; barrier-protective | Epithelial fate (barrier homeostasis maintenance) | RNF186 mediates K48-linked ubiquitination and degradation of substrates such as occludin to maintain epithelial proteostasis. Its loss or the UC-associated A64T mutation leads to protein accumulation, elevated ER stress, increased epithelial apoptosis, impaired barrier integrity, and heightened susceptibility to intestinal inflammation | Occludin (ubiquitination substrate, K48-linked) | IECs | Down-regulated in UC patients | Rnf186-/- | DSS, Oxazolone UC-like | Severe colitis | [48] |
| RING | RNF186 | Genetic susceptibility | - | - | R179X truncation disrupts RNF186 membrane localization and impairs its E3 ligase function, reducing inflammatory cytokine responses to bacterial stimuli | - | - | - | - | - | - | [171] |
| RING | TRAF2 | Imbalance of intestinal immunity | Anti-inflammatory | Innate immune fate (macrophage inflammatory activation threshold) | TRAF2, together with TRAF3 and cIAP, promotes K48-linked ubiquitination and proteasomal degradation of c-Rel and IRF5 in macrophages, thereby limiting TLR-driven cytokine production and protecting against colitis | c-Rel/IRF5 (ubiquitination substrate, K48-linked) | Macrophages (myeloid cells) | - | Traf2fl/fllyz2Cre/+ | DSS | Severe colitis | [79] |
| RING | TRAF3 | Imbalance of intestinal immunity | Anti-inflammatory | Innate immune fate (macrophage inflammatory programming control) | TRAF3 cooperates with TRAF2 and cIAP to promote ubiquitin-dependent degradation of the pro-inflammatory transcription factors c-Rel and IRF5 in macrophages, thereby limiting TLR/IL-1-induced pro-inflammatory cytokine production | c-Rel/IRF5 (ubiquitination substrate) | Macrophages (myeloid cells) | - | Traf3fl/fllyz2Cre/+ | DSS | Severe colitis | [79] |
| RING | TRAF3 | - | - | - | Systemic and mucosal up-regulation (“pre-activation”) of TRAF3 in IBD patients | - | - | Up-regulated in IBD patients | - | - | - | [172] |
| RING | TRAF3 | Defect of intestinal barrier; epithelial inflammatory signaling dysregulation | Anti-inflammatory; IL-17 signaling-restrictive | Epithelial fate (inflammatory signaling-restrictive fate, chemokine production suppression) | TRAF3 negatively regulates IL-17R signaling; NDR1 competitively disrupts TRAF3-IL-17R interaction and facilitates Act1-TRAF6 complex assembly, thereby releasing TRAF3-mediated inhibitory control | - | HeLa; mouse embryonic fibroblasts | - | - | - | - | [173] |
| RING | TRAF4 | Mucosal immune activation | Pro-inflammatory; immune activation-promoting; disease activity-associated | Innate immune fate (UC inflammatory activity aggravation, mucosal inflammation amplification) | TRAF4 promotes immune activation by enhancing NF-κB signaling through GITR and interacting with Msn to activate the JNK pathway | - | - | Up-regulated in plasma of IBD patients | - | - | - | [81] |
| RING | TRAF5 | Imbalance of intestinal immunity | Anti-inflammatory; pathogenic T cell expansion-suppressive | Adaptive immune fate (pathogenic Th cell expansion restraint fate, inflammatory cytokine production suppression) | TRAF5 restrains Th cell-mediated inflammation by limiting NF-κB activation and Th1/Th2/IFN-γ⁺IL-17A⁺ T-cell expansion | - | CD4+ T cells; LPMCs | - | TRAF5-/- | DSS | Severe colitis | [57] |
| RING | TRAF6 | - | Pro-inflammatory; innate immune activation-promoting; inflammatory priming-associated | Innate immune fate (NF-κB/MAPK; cytokine production enhancement) | TRAF6 enhances CD40-mediated NF-κB/JNK/MAPK signaling to promote immune activation | - | - | Up-regulated in plasma, PBMCs, and inflamed colonic mucosa of IBD patients | - | - | - | [81] |
| RING | TRAF6 | Defect of intestinal barrier; imbalance of intestinal immunity | Anti-inflammatory; microbiota-driven inflammation-restrictive; epithelial protective | - | - | - | IECs | - | Traf6IEC-KO | DSS | Exacerbated colitis | [174] |
| RING | TRIM14 | Imbalance of intestinal immunity; chemokine axis dysregulation | Pro-inflammatory; CAC initiation-promoting | Innate immune fate (noncanonical NF-κB activation) | TRIM14 binds NF-κB2 p100/p52 and recruits USP14 to remove K63-linked ubiquitin chains at K332/338/341, thereby blocking p62-dependent selective autophagic degradation of p100/p52, stabilizing p100/p52, enhancing noncanonical NF-κB (RelB/p52, CXCL12/CXCL13) and promoting inflammatory responses | NF-κB2 p100/p52 (TRIM14-USP14-regulated binding partner) | PBMCs; BMDMs; BMDCs; MEFs | - | Trim14-/- | DSS; AOM/DSS | Attenuated colitis; reduced CAC | [82] |
| RING | TRIM21 | Imbalance of intestinal immunity; CD4⁺ T cell-driven mucosal inflammation | Anti-inflammatory; immunosuppressive (Th1/Th17 inhibitory) | Adaptive immune fate (Th1/Th17 differentiation suppression fate, effector T-cell inflammatory program restriction) | TRIM21 inhibits TH1/TH17 differentiation in human IBD CD4+ T cells. TRIM21 deficiency promotes TH1/TH17 differentiation and increases IFN-γ, TNF-α, and IL-17A expression. IRF3 is a downstream target: silencing IRF3 blocks the enhanced TH1/TH17 differentiation in TRIM21⁻/⁻ CD4+ T cells | - | Naive CD4+ T cells | Down-regulated in intestinal mucosa of IBD patients | Trim21-/- | TNBS | Severe colitis | [58] |
| RING | TRIM21 | Imbalance of intestinal immunity; imbalance of intestinal immunity | Anti-inflammatory; CAC initiation-suppressive; epithelial growth-restrictive | Epithelial fate (hyperproliferation suppression, adhesion integrity maintenance) | TRIM21 negatively regulates intestinal epithelial carcinogenesis by restraining epithelial proliferation, maintaining adhesion, limiting tissue remodeling/angiogenesis, and suppressing pro-inflammatory cytokines. Loss of TRIM21 leads to enhanced tumor-promoting inflammation and epithelial transformation | - | - | Down-regulated in intestinal mucosa of IBD and CRC patients | Trim21-/- | AOM/DSS | Severe CAC | [73] |
| RING | TRIM26 | Imbalance of intestinal immunity; TLR-driven innate inflammatory amplification | Pro-inflammatory | Innate immune fate (TLR-dependent inflammatory activation fate, cytokine production enhancement) | TAB1 K11-linked polyubiquitination-dependent TAK1 activation; NF-κB/MAPK signaling amplification; pro-inflammatory cytokine induction | TAB1 (ubiquitination substrate, K11-linked) | PMs, BMDMs, MEFs, THP-1 | - | Trim26-/- | DSS | Attenuated colitis | [50] |
| RING | TRIM27 | Imbalance of intestinal immunity; inflammation-driven tumorigenesis promotion | Pro-inflammatory; CAC initiation-promoting | Epithelial fate; Innate immune fate (cytokine production enhancement fate) | TRIM27 recruits gp130, JAK1 and STAT3 to retromer-positive endosomal structures, facilitates JAK1-STAT3 complex assembly and STAT3 Y705 phosphorylation, amplifying IL-6 signaling cascade | - | HeLa, HT29, RKO | Up-regulated in CRC patients | Trim27-/ | DSS; AOM/DSS | Attenuated colitis; reduced CAC | [74] |
| RING | TRIM27 | Imbalance of intestinal immunity; NOD2 signaling dysregulation | Anti-inflammatory; NOD2 signaling-restrictive | Innate immune fate (PRRs signaling restraint fate, inflammatory cytokine production suppression) | TRIM27 directly binds NOD2 via PRY-SPRY domain and promotes K48-linked ubiquitination and proteasomal degradation of activated NOD2, thereby suppressing MDP-induced NF-κB activation | NOD2 (ubiquitination substrate, K48-linked) | HeLa | Up-regulated in CD patients | - | - | - | [86] |
| RING | TRIM31 | Imbalance of intestinal immunity; NLRP3 inflammasome dysregulation | Inflammasome-suppressive; epithelial barrier-protective (context-dependent) | Epithelial fate (barrier integrity preservation); Innate immune fate (inflammasome suppression) | TRIM31 ubiquitinates NLRP3 (K48-linked) to promote its proteasomal degradation and restrict inflammasome activation; because NLRP3 is protective in DSS-induced mucosal injury, TRIM31 deficiency increases NLRP3 activity and IL-1β/IL-18 maturation | NLRP3 (ubiquitination substrate, K48-linked) | Macrophages; intestinal innate immune cells | - | Trim31-/- | DSS | Attenuated colitis | [51] |
| RING | TRIM58 | Imbalance of intestinal immunity | Anti-inflammatory | Innate immune fate (TLR2 signaling restraint) | TRIM58 associates with TLR2 in myeloid cells and, via its RING-dependent E3 ligase activity, promotes proteasome-dependent degradation of TLR2, thereby terminating TLR2-NF-κB/AP-1 signaling and preventing excessive IL-1β and proinflammatory cyto/chemokine production in DSS colitis | TLR2 (ubiquitination substrate) | Myeloid cells (macrophages/monocytes), IECs | Down-regulated in in UC colonic tissues | Trim58-/-; Trim58MC⁻/⁻ (LysM-Cre) | DSS | Severe colitis | [87] |
| RING | TRIM59 | Imbalance of intestinal immunity; oxidative stress dysregulation | Anti-inflammatory; anti-oxidative stress; anti-apoptotic; epithelial barrier-protective | Epithelial fate (oxidative injury-resistant fate; barrier maintenance) | TRIM59 promotes KEAP1 ubiquitination and degradation, activates NRF2-driven antioxidant signaling, reduces ROS and inflammation, and consequently mitigates colitis progression | KEAP1 (ubiquitination substrate) | IECs | Down-regulated in intestinal mucosa of UC and CD patients | IEC-KOTrim59; IEC-OETrim59 | DSS | Severe colitis (KO); attenuated colitis (OE) | [71] |
| RING | TRIM62 | Imbalance of intestinal immunity; antimicrobial defense-promoting | Pro-inflammatory; antimicrobial defense-promoting | Innate immune fate (CARD9-dependent inflammatory activation fate, cytokine production enhancement) | TRIM62 binds CARD9 and mediates K27-linked polyubiquitination at K125, which is essential for CARD9 activation and downstream proinflammatory cytokine production. The protective C-terminal truncated CARD9 variant fails to interact with TRIM62 and is not ubiquitinated, thereby limiting inflammatory cytokine responses | CARD9 (ubiquitination substrate, K27-linked) | - | - | Trim62-/- | DSS | Severe colitis | [124] |
| RING | TRIM33 | Imbalance of intestinal immunity | Anti-inflammatory; pro-resolution | Innate immune fate (macrophage M2 polarization) | TRIM33 regulates monocyte recruitment and macrophage differentiation, and is required for the M1-to-M2 transition and adequate mTNF expression during inflammatory resolution; its loss leads to persistent inflammation | - | Monocytes, macrophages | Down-regulated in monocytes of CD patients | Trim33-/- | DSS | Severe colitis | [77] |
| RING | TRIM34 | Imbalance of intestinal immunity | Anti-inflammatory; CAC initiation-suppressive | Epithelial fate (goblet cell secretory, mucus barrier maintenance) | TRIM34 in goblet cells regulates TLR signaling-induced Nox/Duox-dependent ROS synthesis, which promotes Muc2 compound exocytosis and enables proper generation of the colonic inner mucus layer. Loss of TRIM34 impairs Muc2 secretion and mucus barrier formation | - | Goblet cells | Down-regulated in colonic mucosa of UC patients | Trim34-/- | DSS; AOM/DSS | Severe colitis; enhanced CAC progression | [75] |
| RING | UHRF1 | Imbalance of intestinal immune tolerance (Treg deficiency-driven inflammation) | Pro-inflammatory; immune tolerance-suppressive | Adaptive immune fate (Treg differentiation) | Uhrf1 sustains DNA methylation of Treg-related genes upon TCR stimulation, thereby limiting Treg differentiation. TGF-β induces Uhrf1 phosphorylation, cytoplasmic sequestration, and degradation, which permits Foxp3 induction and iTreg generation. Uhrf1 loss causes DNA hypomethylation and drives Treg-biased differentiation | - | CD4⁺ T cells (naive T cells → iTreg) | - | Cd4-Cre Uhrf1fl/fl | T cell transfer colitis model | Attenuated colitis | [88] |
| RING | UHRF1 | Imbalance of intestinal immunity; epithelial barrier disruption | Anti-inflammatory; anti-apoptotic; barrier-protective | Epithelial fate (apoptosis); Innate immune fate (inflammatory macrophage activation fate, TNF-α hypersecretion) | Uhrf1 maintains DNA methylation at the Tnf-α promoter in macrophages, thereby restricting TNF-α production and macrophage activation. Uhrf1 deficiency or mutation causes promoter hypomethylation and excessive TNF-α expression, leading to aggravated DSS colitis. TNF-α in turn destabilizes Uhrf1 through ubiquitination-mediated degradation, creating a feed-forward activation loop | - | Macrophages | - | Uhrf1fl/flLyz2-Cre | DSS | Severe colitis | [60] |
Hierarchical regulation of E3-mediated cell fate
As illustrated in Figure 2C, E3 ligases are distributed across epithelial, innate, and adaptive immune compartments, forming an integrated regulatory network that coordinates barrier repair, immune activation, and inflammatory resolution in intestinal inflammation [19, 45] (Figure 2C, Table 4).
The colonic epithelium is composed of multiple specialized intestinal epithelial cell populations, including colonocytes, goblet cells, enteroendocrine cells, and stem cells (Figure 2A). Within IECs, representative E3 ligases such as HACE1, NEDD4L, and RNF186 regulate stress-adaptive responses and barrier-associated fate programs, thereby controlling epithelial survival, regeneration, and injury susceptibility [46-49]. In innate immune cells, E3 ligases, including TRIM26, TRIM31, and Pellino1/3, modulate inflammasome activation and the amplitude of inflammatory signaling, shaping the magnitude and persistence of mucosal immune responses [50-53]. In adaptive immune cells, representative E3 ligases, including ITCH, RNF5, TRAF5, TRIM21, and UHRF1, cooperatively sustain mucosal immune homeostasis by regulating Th1/Th17 differentiation, limiting pro-inflammatory signaling, and stabilizing Treg function, thereby preventing colitis exacerbation and preserving epithelial barrier integrity [54-58]. Together, E3 ligases form a hierarchical, cross-compartment regulatory network that links epithelial repair, innate inflammatory amplitude, and adaptive immune tolerance, thereby coordinating inflammation resolution and tissue protection in intestinal inflammation.
Epithelial cell fate: balancing survival and death to maintain barrier homeostasis
Within an inflammatory microenvironment, intestinal epithelial cells continuously navigate a dynamic balance between survival and death, with E3 ligases acting as central regulators of this decision-making network [59]. TNF-α can trigger epithelial apoptosis; however, UHRF1, HACE1, cIAP1, and RNF5 promote pro-survival programs by catalyzing ubiquitination at key signaling hubs (e.g., RIPK1 and TRAF2), thereby strengthening NF-κB-dependent survival signaling and shifting cell fate toward anti-apoptotic pathways [46, 56, 60, 61]. In contrast, a subset of E3 ligases directly modulates stress-associated death programs: Hrd1 protects the epithelium by alleviating ER stress and limiting inflammatory amplification [62], whereas RNF183 enhances stress-linked apoptotic signaling and exacerbates mucosal injury [63]. Beyond canonical apoptosis control, E3 ligases further shape epithelial fate through multiple adaptive stress-response pathways. In ferroptosis regulation, NEDD4L induces epithelial ferroptosis by suppressing the SLC3A2-GPX4 axis, thereby restricting aberrant proliferation and impeding the transition from inflammation to colorectal cancer [47]. In autophagy control, Parkin promotes autophagy-lysosome-dependent degradation of VDR, dampening vitamin D signaling and compromising barrier homeostasis [64]. Conversely, MARCH8, RNF8, and RNF128 support epithelial stability by driving selective autophagy to eliminate inflammation-associated substrates, thereby restraining inflammatory spread [65-67]. E3 ligases also exert bidirectional control over oxidative stress adaptation. RINCK and RNF31 suppress NRF2-mediated antioxidant programs, increase ROS accumulation, and aggravate epithelial injury [68, 69]. In contrast, Hakai and TRIM59 enhance NRF2-dependent antioxidant and metabolic homeostatic responses to buffer oxidative stress, maintain barrier function, and limit persistent inflammation [70, 71]. In parallel, MARCH3, TRIM21, and TRIM27 target inflammatory receptors and key nodes in innate immune signaling, whereas TRIM34 in goblet cells suppresses feed-forward inflammatory cascades, thereby sustaining the epithelial-immune interface [72-75]. Collectively, the epithelial E3 ligase network integrates apoptosis, autophagy, ferroptosis, and oxidative stress pathways to form a central regulatory hub for epithelial fate decisions under inflammatory stress, thereby shaping barrier homeostasis and influencing disease trajectories.
Innate immune cell fate: inflammasome-driven pyroptosis and functional polarization
During intestinal inflammation, innate immune cells undergo both programmed pyroptosis and functional polarization, and E3 ligases serve as key determinants of these fate transitions. TRIM31 restrains inflammasome activation and pyroptosis by promoting ubiquitination and degradation of NLRP3 [51]. In contrast, RNF31 enhances NLRP3 inflammasome activity via ubiquitin-dependent mechanisms, amplifying inflammatory output and exacerbating tissue damage [76]. In macrophage polarization, Pellino1 drives pathological polarization by augmenting STAT3-dependent inflammatory signaling [52], whereas TRIM33 promotes inflammation resolution by supporting monocyte differentiation and facilitating the M1-to-M2 transition [77]. Moreover, c-Cbl, MARCH3, TRAF2, TRAF3, and UHRF1 collectively restrain excessive cytokine production by tuning signaling thresholds at inflammatory receptors and regulating the NF-κB/IRF transcriptional axis, thereby maintaining mucosal innate immune homeostasis [60, 72, 78, 79]. Conversely, Pellino3, RNF40, TRAF4, TRAF6, TRIM14, and TRIM26 act as signal amplifiers that increase TLR/NOD pathway output [50, 53, 80-82], whereas cIAP1/2, RNF99, RNF138, TRIM27, and TRIM58 impose negative feedback to limit inflammatory signaling strength [83-87]. In summary, these opposing modules define the magnitude of innate immune responses and thereby influence the persistence of intestinal inflammation and the risk of malignant progression. Overall, E3 ligases fine-tune innate immune fate by bidirectionally regulating inflammasome activity, polarization circuits, and receptor-centered inflammatory signaling networks.
Adaptive immune cell fate: lineage specification and immune tolerance
In the adaptive immune compartment, E3 ligases govern T-cell lineage specification and immune tolerance by controlling the stability of transcription factors and the intensity of upstream signaling. ITCH suppresses Th17 differentiation by ubiquitinating and promoting the degradation of RORγt, while also contributing to the Treg program [54, 55]. Loss of ITCH results in excessive Th17 expansion, impaired barrier repair, and immune imbalance, thereby driving spontaneous colitis and increasing cancer risk [54, 55]. UHRF1 exerts cell-type-dependent effects in intestinal inflammation. It limits Treg differentiation by maintaining DNA methylation, and its loss attenuates colitis in T cell transfer models [88]. In contrast, UHRF1 restrains TNF-α expression in macrophages via promoter methylation, and its deficiency aggravates DSS-induced colitis [60]. In addition, RNF5, TRAF5, and TRIM21 cooperatively restrain pathogenic Th1/Th17 effector programs and limit excessive adaptive immune activation; deficiency in these regulators exacerbates CD4⁺ T-cell-driven colitis phenotypes [56-58].
Tissue-level regulation of disease outcomes: from inflammatory homeostasis to a cancer-permissive state
At the tissue level, E3 ligases coordinate epithelial homeostasis with innate and adaptive immune responses, thereby critically influencing whether inflamed colonic tissue resolves back to homeostasis or progresses into chronic inflammation. This tissue-scale decision subsequently shapes the long-term risk of CAC. Several E3 ligases, including MARCH3, RNF128, RNF138, and TRIM21, inhibit the colitis-to-cancer transition by suppressing IL-6-STAT3 signaling, constraining aberrant epithelial proliferation, and/or dampening NF-κB activity [72, 73, 85, 89]. In contrast, TRIM27 promotes this process [74]. Clinical observations further support this directional regulation, with the former group frequently downregulated in IBD or CRC tissues [72, 73, 85, 89], whereas TRIM27 is markedly upregulated in CRC specimens, consistent with their opposing functions along the inflammation-tumor fate axis [74].
It should be noted that most of the regulatory mechanisms summarized in this section are derived from inflammation-driven experimental systems, including chemically induced models such as azoxymethane/dextran sulfate sodium (AOM/DSS) and genetically engineered models of chronic colitis [47, 90]. These models primarily recapitulate key pathological features of CAC, including persistent inflammatory stress, epithelial barrier disruption, immune microenvironment remodeling, and inflammation-dependent tumor initiation and progression. In contrast, sporadic colorectal cancer is predominantly driven by oncogenic mutations and develops within a distinct etiological and microenvironmental context. Therefore, while certain ubiquitin-dependent regulatory pathways may be shared, caution should be exercised when extrapolating inflammation-centered mechanisms directly to mutation-driven sporadic CRC.
In parallel, TRIM34, HACE1, ITCH, and NEDD4L restrict the establishment of a pro-tumorigenic premalignant microenvironment by maintaining epithelial stability and/or restraining excessive immune activation [46, 47, 54, 75]. Conversely, Pellino1, Pellino3, and TRIM14 promote chronic inflammatory maintenance and amplify tumor-promoting signaling cascades [52, 53, 82]. Collectively, these antagonistic E3 ligases form a dynamic regulatory network at the tissue scale, orchestrating the balance between inflammatory resolution and cancer-permissive transformation.
Fate “integrators” across cell types and signaling layers: ITCH and RNF186
A subset of E3 ligases exerts coordinated control across multiple cell types and signaling layers, functioning as fate “integrators.” Here, ITCH and RNF186 exemplify this role. ITCH coordinately regulates adaptive immunity, epithelial repair, and fibrotic fate by promoting degradation of RORγt and HIC-5, thereby balancing Th17 differentiation, epithelial homeostasis, and fibroblast activation, ultimately constraining inflammation-associated carcinogenesis and intestinal fibrosis [54, 55, 90]. RNF186 plays bidirectional roles in intestinal inflammation: it can promote ER stress-associated apoptosis via BNip1 ubiquitination [91], yet it also preserves mucosal homeostasis by supporting EPHB2-dependent autophagy, ATF6-UPR signaling, and Occludin-mediated tight-junction proteostasis [48, 49, 92]; loss-of-function variants compromise these protective arms and increase colitis susceptibility [48, 49, 92].
Together, the hierarchical regulation of cell fate by E3 ubiquitin ligases provides a unifying framework linking inflammation to malignant progression. At the cellular level, these ligases dictate epithelial survival versus death, macrophage pyroptosis versus polarization, and T-cell effector versus regulatory differentiation. At the tissue level, these micro-scale fate choices are integrated into macro-scale outcomes, determining whether inflammation resolves or persists (Figure 4A). Dysregulated E3 activity disrupts this equilibrium, driving immunopathology and/or promoting tumorigenesis. Thus, the ubiquitin system not only controls protein turnover but, through cross-scale regulatory networks, ultimately shapes the clinical fate of colonic tissue—healing, chronic inflammation, or malignant transformation.
Hierarchical regulation of cell fate by DUBs
DUBs counterbalance ubiquitin-dependent signaling to fine-tune inflammatory responses in the gut [93, 94]. By removing ubiquitin chains from key regulators, DUBs modulate protein stability, activity, and localization, thereby shaping the behavior of epithelial and immune cells under stress. Rather than merely buffering signals, DUBs construct multilayered regulatory circuits that restore signaling thresholds, resolve inflammation, and preserve cellular equilibrium. In intestinal inflammation, they orchestrate fate decisions across epithelial, innate, and adaptive immune compartments, ultimately impacting whether tissue regains homeostasis or progresses toward chronic inflammation and malignancy. DUBs fall into four major families—USP, OTU, JAMM, and MJD, each defined by distinct catalytic architectures and chain-linkage specificities [94-96] (Figure 2D, Figure 4B, Table 5).
Roles of deubiquitinating enzymes in intestinal inflammation and tumorigenesis.
| Family | Gene | Pathogenesis of IBD/CAC | Effect | Cell fate | Mechanism/major finding | Target protein classification | Cell type | Alteration in patients | Transgenic mice | Disease model | Disease Phenotype | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MJD | JOSD2 | Imbalance of intestinal immunity | Anti-inflammatory; barrier-protective; CAC initiation-suppressive | Epithelial fate (apoptosis and goblet cell loss); Innate immune fate (macrophage inflammatory activation) | JOSD2 deubiquitinates K63-linked chains on IMPDH2 at K134, suppressing IMPDH2 activity and NF-κB-mediated inflammatory signaling in macrophages, thereby limiting colitis and CAC | IMPDH2 (deubiquitination substrate, K63-linked) | Macrophages (myeloid cells) | Up-regulated in colonic macrophages from UC and CD patients | Josd2-/-; AAV6-JOSD2 | DSS; AOM/DSS | Severe colitis; enhanced CAC progression | [108] |
| OTU | TNFAIP3 (A20) | Defect of intestinal barrier; imbalance of intestinal immunity | Pro-inflammatory (IEC context-dependent); apoptosis-promoting | Epithelial fate (RIPK1-dependent apoptosis induction, ripoptosome enhancement, barrier breakdown) | IEC-specific A20 overexpression sensitizes epithelial cells to TNF-induced, RIPK1-dependent apoptosis by promoting Ripoptosome assembly and caspase-8/3 activation, resulting in epithelial barrier breakdown and acute inflammation | - | IECs | Up-regulated in UC and CD patients | villin-A20 transgenic (overexpression) | TNF injection model | Severe colitis | [103] |
| OTU | TNFAIP3 (A20) | Imbalance of intestinal immunity | Anti-inflammatory; immune homeostasis-maintaining | Innate immune fate (DC activation restraint, cytokine overproduction suppression); Adaptive immune fate (T cell activation limitation, peripheral tolerance maintenance) | A20 restricts MyD88-dependent and MyD88-independent signals in dendritic cells, limiting NF-κB activation, co-stimulatory molecule upregulation, and IL-6/TNF production, thereby preventing aberrant activation and expansion of T cells and maintaining intestinal immune homeostasis | - | Dendritic cells (CD11c⁺ DCs) | - | A20-/-, A20fl/fl Cd11c-Cre | DSS; spontaneous colitis | Severe colitis | [107] |
| OTU | TNFAIP3 (A20) | Imbalance of intestinal immunity | Anti-inflammatory; immune homeostasis-restoring | Epithelial fate (inflammatory signaling attenuation, tissue damage reduction); Adaptive immune fate (Th17 suppression, Treg enhancement) | A20 overexpression in intestinal epithelial cells inhibits IL-6- and LPS-induced STAT3 and NF-κB activation, decreases colonic IL-17, IL-1β and TNF-α expression, and shifts the Th17/Treg balance, thereby ameliorating colitis | - | HT29 | Down-regulated in colonic mucosa from UC patients | A20-overexpressing (plasmid-treated) | DSS | Attenuated colitis | [121] |
| OTU | TNFAIP3 (A20) | Defect of intestinal barrier; imbalance of intestinal immunity | Anti-inflammatory; anti-apoptotic; epithelial protective | Epithelial fate (TNF-induced apoptosis suppression, epithelial survival maintenance, barrier integrity preservation) | A20 protects intestinal epithelial cells by inhibiting TNF-induced apoptosis; A20 deficiency causes TNF-driven epithelial barrier breakdown, commensal bacterial translocation, systemic inflammation, and heightened susceptibility to colitis | - | IECs | - | A20IEC-KO | DSS | Severe colitis | [104] |
| JAMM | BRCC3 | Imbalance of intestinal immunity | Pro-inflammatory; inflammasome-activating | Innate immune fate (macrophage inflammasome activation; IL-1β and IL-18 production) | BRCC3-mediated deubiquitination of NLRP3 LRR promotes NLRP3 oligomerization and inflammasome activation | NLRP3 (deubiquitination substrate, K63-linked) | Macrophages (BMDMs, THP-1-derived macrophages) | - | - | - | - | [111] |
| OTU | CYLD | Defect of intestinal barrier; imbalance of intestinal immunity | Pro-inflammatory (context-dependent); necroptosis-promoting | Epithelial fate (RIP3-mediated necroptosis induction, Paneth cell loss, barrier disruption) | CYLD deubiquitinase activity promotes RIP1/RIP3-dependent necroptosis downstream of TNFR1; inhibition of CYLD catalytic activity in IECs (CYLDΔ932IEC) prevents RIP3-mediated epithelial necrosis and spontaneous colitis in FADDIEC-KO mice | - | IECs | - | CYLDΔ932IEC, FADDIEC-KO | Spontaneous microbiota-dependent colitis in FADDIEC-KO mice | Attenuated colitis | [175] |
| OTU | CYLD | Imbalance of intestinal immunity | Anti-inflammatory; CAC initiation-suppressive | Epithelial fate (inflammation-driven survival signaling enhancement); Innate immune fate (NF-κB/JNK hyperactivation, cytokine overproduction) | CYLD removes K63-linked ubiquitin from TRAF2 and NEMO to restrain NF-κB/JNK signaling. CYLD loss leads to TRAF2/NEMO hyperubiquitination, sustained NF-κB/JNK activation, and increased CAC | TRAF2 and NEMO (deubiquitination substrate, K63-linked) | B cells, T cells, macrophages | - | Cyld-/- | DSS; DSS-induced CAC | Severe colitis; prone to CAC | [109] |
| OTU | CYLD | Defect of intestinal barrier; imbalance of intestinal immunity | Anti-inflammatory; inflammasome-suppressive | Epithelial fate (barrier integrity maintenance, microbial translocation limitation); Innate immune fate (inflammasome activation restraint, IL-18 maturation control) | CYLD deubiquitinates NLRP6 to restrict K63-linked ubiquitination, limiting NLRP6-ASC complex formation, caspase-1 activation, and IL-18 maturation, thereby preventing excessive intestinal inflammation | NLRP6 (deubiquitination substrate, K63-linked) | IECs | - | Cyld-/-; IEC-CyldΔ9 | C. rodentium-induced colitis; TNBS | Severe colitis | [105] |
| OTU | CYLD | Imbalance of intestinal immunity | Anti-inflammatory; innate immune signaling-terminating | Innate immune fate (TLR-NF-κB/MAPK termination, cytokine overproduction suppression) | CYLD deubiquitinates TRAF6 to terminate TLR-triggered NF-κB signaling; in this study GIT2 recruits CYLD to TRAF6 and enhances CYLD-mediated deubiquitination of TRAF6, thereby limiting TLR-induced NF-κB and MAPK activation | TRAF6 and NEMO (deubiquitination substrate, K63-linked) | BMDMs | - | - | - | - | [125] |
| OTU | CYLD (short splice variant, sCYLD) | Imbalance of intestinal immunity | Pro-inflammatory; immune tolerance-disruptive; TGF-β signaling-suppressive | Adaptive immune fate (Treg/Th17 differentiation inhibition, Th1 polarization enhancement, effector memory T cell expansion) | sCYLD promotes K63-linked ubiquitination and nuclear translocation of SMAD7 in CD4⁺ T cells, recruiting SMAD7 into SMAD3/4 DNA-binding complexes to inhibit TGF-β signaling, thereby impairing Treg and Th17 differentiation, enhancing Th1 effector responses, and driving colitis | SMAD7 (deubiquitination substrate, K63-linked) | CD4⁺ T cells | Up-regulated in colonic lamina propria T cells from CD patients. | sCYLD/SMAD7 | Spontaneous colitis | Severe colitis | [120] |
| OTU | OTUD1 | Imbalance of intestinal immunity | Anti-inflammatory; barrier-protective | Innate immune fate (macrophage inflammatory activation; NF-κB-dependent TNF-α/IL-6/IL-1β production) | OTUD1 binds RIPK1 and removes K63-linked ubiquitin chains (notably at K627), blocking NEMO recruitment and RIPK1-mediated NF-κB activation, thereby limiting proinflammatory cytokine production | RIPK1 (deubiquitination substrate, K63-linked) | Hematopoietic cells | Down-regulated in intestinal mucosa from UC patients | Otud1-/- | DSS | Susceptibility to colitis | [126] |
| OTU | OTUD5 | Imbalance of intestinal immunity | Pro-inflammatory | Innate immune fate (APC/macrophage inflammatory activation; TNF-α production); Adaptive immune fate (Th17 differentiation regulation; context-dependent) | OTUD5 is induced by IFN-γ via p38/MAPK in intestinal lamina propria antigen-presenting cells and sustains the inflammatory cytokine response, as antisense-mediated OTUD5 knockdown in IBD and TNBS-colitis LPMCs reduces p38 activation and TNF-α expression | - | Lamina propria antigen-presenting cells and epithelial cells | Up-regulated in inflamed ileal and colonic mucosa from UC and CD patients | - | TNBS | - | [112] |
| OTU | OTUD6A | Imbalance of intestinal immunity | Pro-inflammatory; inflammasome-activating; CAC initiation-promoting | Innate immune fate (macrophage NLRP3 inflammasome activation; IL-1β/IL-18 maturation and pyroptosis) | OTUD6A binds NLRP3 and removes K48-linked ubiquitin chains at K430 and K689, stabilizing NLRP3 and enhancing NLRP3 inflammasome activation and IL-1β production in macrophages | NLRP3 (deubiquitination substrate, K48-linked) | Macrophages (BMDMs; myeloid cells) | Up-regulated in colonic mucosa from UC patients | Otud6a-/- | DSS; TNBS; AOM/DSS | Attenuated colitis; reduced CAC | [113] |
| OTU | OTULIN | Defect of intestinal barrier; imbalance of intestinal immunity | Anti-inflammatory; anti-apoptotic; barrier-protective | Epithelial fate (TNF-induced apoptosis, epithelial destruction, barrier disruption) | OTULIN deficiency impairs TNFR1 complex I formation and LUBAC recruitment upon TNF stimulation, promotes formation of cytosolic death-inducing complex II (FADD-caspase-8), leading to excessive TNF-induced epithelial apoptosis and barrier breakdown | - | IECs | - | OTULINIEC-KO | DSS | Susceptibility to colitis | [97] |
| USP | USP1 | Genetic susceptibility | - | - | SNP (rs1748195) in USP1 gene is associated with CD risk | - | - | - | - | - | [176] | |
| USP | USP3 | Genetic susceptibility | - | - | Polymorphisms in USP3 genes are associated with both CD and UC patients | - | - | - | - | - | [45] | |
| USP | USP4, USP40 | Genetic susceptibility | - | - | Polymorphisms in USP4 and USP40 genes are associated with CD and UC patients | - | - | - | - | - | [21, 45, 177] | |
| USP | USP5, USP15, USP19, USP39 | Genetic susceptibility | - | - | Polymorphisms in USP5, 15, 18, 39 genes are associated with UC patients | - | - | - | - | - | [177] | |
| USP | USP7 | Defect of intestinal barrier; imbalance of intestinal immunity | Pro-inflammatory; pro-oxidative | Epithelial fate (barrier disruption, epithelial injury | USP7 deubiquitinates and stabilizes AMBRA1, which suppresses NRF2 antioxidant signaling and increases oxidative stress in IECs; USP7 inhibition reduces AMBRA1, restores NRF2 activity, and alleviates colitis | AMBRA1 (deubiquitination substrate, K48-linked) | IECs | - | USP7 inhibitor (P5091) | DSS | Attenuated colitis | [102] |
| USP | USP7 | Imbalance of intestinal immunity; impaired Treg stability | Anti-inflammatory; maintains Treg suppressive function | Adaptive immune fate (Foxp3 stabilization, Treg suppressive function maintenance) | USP7 deubiquitinates and stabilizes Foxp3, enhancing Treg suppressive function; USP7 inhibition or knockdown reduces Foxp3 and abolishes Treg-mediated resolution of colitis | Foxp3 (deubiquitination substrate) | Treg cells | - | Foxp3-GFP mice; Treg with shUSP7 | Adoptive-transfer colitis | - | [116] |
| USP | USP8 | Imbalance of intestinal immunity | Anti-inflammatory; immune homeostasis-maintaining | Adaptive immune fate (thymocyte maturation, IL-7Rα expression, Treg suppressive function) | USP8 acts in the TCR signalosome and supports Foxo1-dependent IL-7Rα expression, maintaining T-cell development, homeostasis and Treg function; its loss disrupts these programs and permits expansion of colitogenic γδ T cells | Gads and 14-3-3β (deubiquitination substrate) | Thymocytes and peripheral T cells | - | Usp8f/fCd4-Cre; Usp8f/fCd4-CreERT2 | Spontaneous colitis | Severe colitis | [117] |
| USP | USP9X | Defect of intestinal barrier | Anti-inflammatory; CAC initiation-suppressive | Epithelial fate (crypt progenitor proliferation control, goblet/Paneth cell differentiation, epithelial regeneration) | USP9X binds FBW7α/β and removes degradative K48-linked polyubiquitin chains, stabilizing FBW7 and thereby promoting degradation of SCF(FBW7) substrates (c-MYC, NICD1, c-JUN, cyclin E); intestinal Usp9x loss lowers Fbw7, increases these oncoproteins | FBW7 (deubiquitination substrate, K48-linked) | IECs | - | Usp9xfl/flVillin-Cre | DSS; AOM/DSS | Severe colitis; prone to CAC | [98] |
| USP | USP13 | Defect of intestinal barrier; ER stress dysregulation | Anti-inflammatory; anti-apoptotic; stress-adaptive; barrier-protective | Epithelial fate (IEC apoptosis inhibition, barrier integrity maintenance) | USP13 in intestinal epithelial cells binds GRP78 and removes K63-linked ubiquitin at K327 via its catalytic C343 site, suppressing GRP78-mediated ER stress and apoptosis and maintaining tight-junction-dependent barrier integrity during DSS colitis | GRP78 (deubiquitination substrate, K63-linked) | IECs | Down-regulated in inflamed colonic/rectal mucosa of IBD patients | Usp13IEC-KO; AAV-Usp13 | DSS | Severe colitis (Usp13IEC-KO); attenuated colitis (AAV-Usp13) | [25] |
| USP | USP16 | Imbalance of intestinal immunity | Pro-inflammatory; CAC initiation-promoting | Innate immune fate (macrophage activation, cytokine and chemokine production) | USP16 directly binds IKKβ and selectively removes K33-linked polyubiquitin chains from IKKβ at lysine 238, facilitating IKKβ interaction with p105, enhancing p105 phosphorylation and processing to p50, thereby selectively amplifying canonical NF-κB signaling without affecting IκBα phosphorylation | IKKβ (deubiquitination substrate, K33-linked) | Macrophages; BMDMs; myeloid cells | Up-regulated in colonic macrophages and inflamed mucosa of UC and CD patients | Usp16fl/flLyz2-Cre+ | DSS; AOM/DSS | Attenuated colitis; reduced CAC | [114] |
| USP | USP21 | Imbalance of intestinal immunity | Anti-inflammatory (via Th17 suppression); immune differentiation-restrictive | Adaptive immune fate (Th17 differentiation inhibition, CD4⁺ T cell lineage regulation) | USP21 interacts with AhR and removes K48-linked polyubiquitin chains at K432, stabilizing AhR but suppressing its transcriptional activity. Through this deubiquitination-dependent inhibition of AhR, USP21 negatively regulates Th17 differentiation | AhR (deubiquitination substrate, K48-linked) | CD4+ T cell; Th17 cells | - | - | - | - | [118] |
| USP | USP22 | Defect of intestinal barrier; imbalance of intestinal immunity | Anti-inflammatory; CAC initiation-suppressive; epigenetic transcription-repressive | Epithelial fate (epithelial damage aggravation, barrier impairment); Innate immune fate (cytokine overproduction, immune cell infiltration) | USP22 epigenetically suppresses SPARC transcription by removing monoubiquitinated H2B (H2Bub1) and reducing H3K27ac occupancy at the SPARC promoter and gene body, thereby limiting SPARC expression and restraining inflammation-associated gene programs | H2Bub1 (deubiquitination substrate) | IECs; HCT116 | Down-regulated in UC patients with neoplasia | Usp22fl/fl Villin-CreERT2 | DSS; DSS (Apc1638N/+ CAC model) | Severe colitis; prone to CAC | [99] |
| USP | USP25 | Defect of intestinal barrier; imbalance of intestinal immunity | Anti-inflammatory; barrier-protective | Epithelial fate (tight junction maintenance, epithelial regeneration, barrier integrity preservation) | USP25 interacts with phospho-STAT3 and catalyzes K48-linked deubiquitination of p-STAT3Y705 at K409, preventing its proteasomal degradation and maintaining STAT3 activity to promote IL-10/IL-22 and tight junction protein expression in intestinal epithelial cells | STAT3 (deubiquitination substrate, K48-linked) | IECs | Down-regulated in colonic mucosa of UC patients | Usp25-/-; AAV8-Usp25 | DSS | Severe colitis | [100] |
| USP | USP25 | Genetic susceptibility | - | - | USP25 locus shows genome-wide significant association with IBD in African Americans; rs7278277 near USP25 is a GWS SNP for IBD | - | - | - | - | - | - | [24] |
| USP | USP25 | Defect of intestinal barrier; imbalance of intestinal immunity | Pro-inflammatory (infection context); epithelial proliferative signaling-promoting; CAC initiation-promoting | Epithelial fate (secretory lineage regulation, proliferative signaling enhancement); Innate immune fate (suppressed antibacterial responses, cytokine remodeling) | USP25 deubiquitinates and stabilizes TRAF3 to restrain TLR-triggered signaling; in the intestine, USP25 in non-hematopoietic cells limits antibacterial cytokines/antimicrobial peptides and, by promoting Wnt signaling and restraining SOCS3-pSTAT3, supports colitis-associated tumorigenesis, whereas USP25 deficiency or inhibition enhances antibacterial immunity | - | ECs; LPMCs; secretory cells | - | Usp25-/- | DSS; AOM/DSS | Attenuated colitis; reduced CAC | [115] |
| USP | USP28 | Imbalance of intestinal immunity | Anti-inflammatory; immune homeostasis-maintaining | Adaptive immune fate (T cell activation control, Th17 differentiation modulation, enhanced Treg suppressive function, IL22 overproduction) | USP28 in T cells restrains STAT5 phosphorylation and IL-22 production; Usp28-/- T cells show enhanced STAT5 activation, increased IL-22/IFN-γ and altered Th17/Treg functions, leading to greater susceptibility to DSS-induced colitis | - | T lymphocytes (CD4⁺ Th17/Treg/iTreg and CD8⁺ T cells) | - | Usp28-/- | DSS (acute/chronic) | Severe colitis | [119] |
| USP | USP38 | Imbalance of intestinal immunity | Anti-inflammatory; transcription-repressive | Innate immune fate (macrophage/DC inflammatory activation, IL-6 and IL-23a overproduction) | USP38 deubiquitinates H2B K120 and recruits/stabilizes KDM5B to remove H3K4me3 at Il6 and Il23a promoters, thereby limiting NF-κB (p65, c-Rel, p50) binding and selectively suppressing IL-6 and IL-23α expression | KDM5B (deubiquitination substrate, K48-linked) | Macrophages (BMDMs, peritoneal macrophages); BMDCs | - | Usp38-/- | DSS | Severe colitis | [110] |
| USP | USP44 | Genetic susceptibility | - | - | USP44 promoter hypermethylation is part of a 5-gene methylation panel that is enriched in neoplastic and non-neoplastic colonic mucosa from IBD patients with dysplasia/cancer and from high-risk IBD patients, serving as an early biomarker of IBD-associated colorectal neoplasia | - | Colonic mucosal biopsies | Hypermethylated in IBD mucosa | - | - | - | [178] |
| USP | USP47 | Defect of intestinal barrier; imbalance of intestinal immunity | Anti-inflammatory; epithelial protective | Epithelial fate (NF-κB activation control, epithelial injury attenuation) | USP47 binds TRAF6 in intestinal epithelial cells and removes K63-linked ubiquitin chains (notably at Lys124), thereby restraining TRAF6-dependent NF-κB activation and epithelial inflammatory cytokine production and protecting against colitis | TRAF6 (deubiquitination substrate, K63-linked) | IECs | Down-regulated in colonic mucosa of UC and CD patients | Usp47-/- | DSS | Severe colitis | [101] |
Hierarchical regulation of DUB-mediated cell fate
As shown in Figure 2D and Table 5, DUBs are distributed across intestinal epithelial, innate immune, and adaptive immune compartments, forming a cross-cell type regulatory network that coordinately balances inflammatory responses and tissue repair.
In IECs, OTULIN, USP9X, USP13, USP22, USP25, and USP47 maintain barrier homeostasis and attenuate colitis progression by suppressing NF-κB, STAT3, and inflammation-related signaling pathways [25, 97-101]. In contrast, USP7 enhances oxidative stress and exacerbates inflammatory responses under specific pathological conditions [102], whereas A20 and CYLD exhibit context-dependent dual regulatory roles [103-106]. In innate immune cells, JOSD2, CYLD, A20, OTUD1, and USP38 exert anti-inflammatory effects by limiting inflammatory signal amplification and reducing tissue damage [107-110]. By comparison, BRCC3, OTUD5, OTUD6A, USP16, and USP25 promote immune activation through enhanced cytokine production, thereby modulating the magnitude of inflammatory responses [111-115]. In adaptive immune cells, CYLD, USP7, USP8, USP21, and USP28 regulate T cell activation and the Th17/Treg differentiation balance, maintaining mucosal immune homeostasis and restraining excessive inflammation [109, 116-119]. Conversely, sCYLD promotes inflammatory signaling activation in T cells and enhances pro-inflammatory responses [120]. Collectively, DUBs establish a compartmentalized and reversible deubiquitination regulatory system that cooperates with E3 ligases to coordinately orchestrate inflammation resolution and tissue protection in the intestine.
Epithelial cell fate: balancing apoptosis and stress adaptation to maintain barrier integrity
DUBs preserve epithelial integrity by modulating apoptosis, endoplasmic reticulum (ER) stress, and oxidative responses. OTULIN and USP13 stabilize the epithelial barrier by inhibiting inflammatory signaling (e.g., NF-κB) or alleviating ER stress, thereby limiting TNF-induced or stress-driven epithelial apoptosis [25, 97]. USP25 and USP13 act as anti-stress protectors by either dampening UPR signaling or sustaining STAT3 activation, contributing to epithelial cell survival and tissue repair [25, 100]. In contrast, USP7 suppresses NRF2-dependent antioxidant pathways, resulting in ROS accumulation and increased vulnerability to oxidative injury [102]. These enzymes collectively constitute a DUB network that calibrates epithelial stress responses and determines survival versus death outcomes under inflammatory challenge.
Innate immune cell fate: amplification versus suppression of inflammatory programs
DUBs determine the amplitude and persistence of inflammatory responses in innate immune cells such as macrophages. BRCC3 and OTUD6A enhance inflammasome activation by removing ubiquitin modifications from NLRP3, thereby promoting IL-1β/IL-18 secretion and pyroptosis, which amplifies inflammatory output [111, 113]. USP16 reinforces pro-inflammatory activation by augmenting IKKβ-p105 signaling in macrophages, driving sustained inflammation [114]. In contrast, USP38 suppresses immune overactivation by deubiquitinating H2B and stabilizing KDM5B, which limits NF-κB-dependent IL-6 and IL-23 expression [110]. Taken together, these DUBs form a regulatory module that tunes the polarization state of innate immune cells and dictates inflammatory outcomes.
Adaptive immune cell fate: T cell differentiation, homeostasis, and effector balance
In the adaptive immune system, DUBs regulate the fate of T cell subsets such as Tregs and Th17 cells by modulating activation thresholds and maintaining transcription factor stability. OTUD5 promotes Th17 differentiation and sustains its pro-inflammatory effector functions [112]. In contrast, USP8 stabilizes Tregs by enhancing the Foxo1-IL-7Rα signaling axis [117], and USP21 inhibits Th17 differentiation by constraining AhR activity [118]. These DUBs collectively act within the inflammatory milieu to uphold immune tolerance and prevent dysregulated adaptive immune activation.
Tissue-level outcomes: inflammatory resolution versus tumor-permissive remodeling
At the tissue scale, DUBs integrate epithelial homeostasis, immune responses, and signaling adaptation to shape inflammatory outcomes, determining whether inflamed tissue resolves toward mucosal homeostasis or evolves into a tumor-permissive state. OTUD6A and USP16 promote chronic inflammatory persistence and CAC development by enhancing NLRP3 inflammasome activation or IKKβ-NF-κB signaling [113, 114]. USP25 compromises mucosal antimicrobial defense and epithelial secretory lineage integrity by modulating secretory cell development and the Wnt pathway, thereby facilitating tumorigenesis [115]. In contrast, JOSD2 attenuates tissue inflammatory burden by suppressing macrophage inflammatory pathways [108]; CYLD limits NF-κB and JNK activation [106], USP9X supports epithelial stem/progenitor cell differentiation [98], and USP22 mitigates chronic epithelial injury through transcriptional repression [99]. Collectively, these DUBs define opposing tissue-level outcomes, ranging from stable mucosal homeostasis to inflammation-driven carcinogenic remodeling.
Notably, A20 and CYLD exemplify the context-dependent dual roles of DUBs across distinct cellular environments. Deletion of A20 results in uncontrolled NF-κB activation and systemic inflammation [107, 121]. In contrast, in IECs, A20 exhibits a dose-dependent bifunctional role in TNF signaling [103, 104]. Loss of A20 sensitizes epithelial cells to TNF-induced apoptosis by impairing NF-κB-dependent survival programs [104], whereas excessive A20 expression paradoxically promotes RIPK1-dependent ripoptosome assembly and caspase-8 activation [103]. Mechanistically, both insufficient and excessive A20 activity destabilize TNFR1 signaling homeostasis by altering the balance between membrane-associated complex I and cytosolic death-inducing complex II formation. This threshold-dependent regulatory behavior positions A20 as a rheostat rather than a unidirectional suppressor of inflammation in epithelial compartments. CYLD exhibits similar complexity: in IECs, it promotes necroptosis and epithelial barrier disruption [106], while in immune cells, it exerts anti-inflammatory effects by restricting TRAF-NEMO signaling and NLRP6 inflammasome activation [105, 109]. The short splice variant sCYLD further exacerbates inflammation by inhibiting TGF-β signaling, destabilizing Treg cells, and enhancing Th1/Th17 responses [120].
Collectively, DUBs fine-tune ubiquitination landscapes to orchestrate hierarchical fate decisions within the inflammatory microenvironment. By controlling cell survival, immune balance, and tissue pathology, DUBs serve as critical nodes linking molecular signaling dynamics to disease outcomes, including resolution, chronic inflammation, or tumorigenesis.
From cell fate modulation to pathway control: multi-layered roles of UMEs in inflammatory signaling networks
Following recognition of the layered role of UMEs in shaping cell-fate decisions, accumulating evidence reveals their equally critical function in precisely modulating key molecular nodes within well-defined inflammatory signaling pathways (Figure 5, Tables 3-5). In intestinal inflammation, UMEs dynamically regulate signal transduction complexes, modulate adaptor protein activity, and control the stability of transcription factors across core axes such as TLRs, TNFRs, NOD-like receptors, and STAT signaling, thereby shaping the amplitude, duration, and outcomes of immune responses.
Multi-layered regulation of inflammatory signaling networks by UMEs in intestinal inflammation. Schematic overview illustrating how ubiquitin-modifying enzymes (UMEs), including E2 conjugating enzymes, E3 ligases, and DUBs, coordinate the regulation of key inflammatory signaling pathways in intestinal epithelial and immune cells. UMEs modulate receptor-proximal signalosome assembly (TLRs, TNFR, CLRs, and NOD2), downstream kinase cascades (TAK1-TAB, MAPKs, and IKK complexes), transcriptional programs (NF-κB, AP-1, STAT3), stress-responsive pathways (ER stress and oxidative stress signaling), and inflammasome activation (NLRP3/NLRP6). These coordinated regulatory layers collectively shape cell fate outcomes, including inflammatory activation, stress adaptation, programmed cell death, and barrier homeostasis. Blue boxes indicate E3 ligases, red boxes indicate DUBs, and green boxes represent E2 enzymes. Solid arrows denote signaling flow, whereas dashed arrows indicate transcriptional regulation. Created in BioRender. Qian, C. (2026) https://BioRender.com/a9pj9u4.
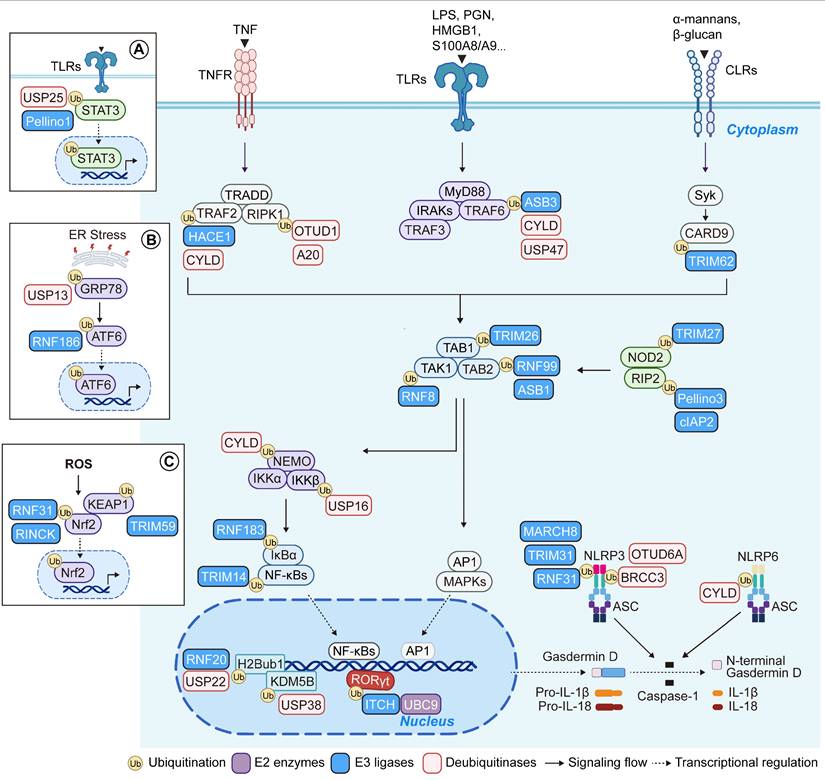
Pattern recognition receptors (PRRs), including TLRs, CLRs, and NOD2, detect microbial and damage-associated molecular patterns and rapidly recruit adaptor proteins such as TRADD, MyD88, IRAKs, and RIP2 to form signaling complexes. These complexes are finely regulated by various E3 ligases (such as Pellino3, ASB3, TRIM62, and cIAP2) and DUBs (including CYLD, USP47, A20, and OTUD1), which together orchestrate signal amplification or termination to balance pro-inflammatory responses with negative feedback control [83, 101, 103, 109, 122-126]. Signals from diverse receptors converge on the TAK1-TAB complex, a key signaling hub whose assembly and activation are modulated by E3 ligases including TRIM26, RNF8, RNF99, and ASB1, thereby allowing graded control over NF-κB and MAPK activation and transcriptional responses [50, 66, 84, 127].
TLR signaling also intersects with STAT3 to influence epithelial-immune crosstalk. Pellino1 promotes STAT3 ubiquitination and nuclear translocation, while USP25 stabilizes STAT3 via deubiquitination, linking inflammatory signaling with epithelial survival [52, 100]. Beyond canonical immune pathways, stress-related signaling is also tightly regulated by the ubiquitin system. Under endoplasmic reticulum stress, RNF186 and USP13 modulate the ubiquitination of GRP78 and ATF6, respectively, adjusting the unfolded protein response and gene expression [25, 92]. In the context of reactive oxygen species (ROS) accumulation, RNF31 and RINCK suppress NRF2 activation, thereby promoting oxidative damage and inflammation [68, 69]. Conversely, TRIM59 enhances KEAP1 degradation, facilitates NRF2 nuclear translocation, and upregulates antioxidant gene expression, mitigating epithelial injury and restraining colitis progression [71]. At the transcriptional level, RNF20, USP22, and USP38 converge on epigenetic regulation to repress inflammatory gene programs [99, 110, 128]. UMEs also govern inflammasome activity: E3 ligases such as RNF31, TRIM31, and MARCH8, along with DUBs like BRCC3, OTUD6A, and CYLD, modulate NLRP3 and NLRP6 ubiquitination, thereby influencing caspase-1 activation, Gasdermin D cleavage, and IL-1β/IL-18 maturation, which shape pyroptotic responses and inflammatory amplification [51, 65, 76, 105, 111, 113]. In adaptive immunity, ITCH suppresses Th17 differentiation by ubiquitinating and degrading RORγt; its deficiency leads to exaggerated NF-κB activity and spontaneous colitis. In parallel, hypoxia represses UBC9 transcription, reducing RORγt SUMOylation and enhancing IL-17 expression, thereby reinforcing Th17-driven pathogenicity [41, 55]. Altogether, UMEs operate across multiple layers of the intestinal inflammatory network, ranging from signalosome assembly to transcriptional control, to define the trajectory of immune activation, tissue injury, and homeostatic resolution, making them pivotal regulators of inflammation and disease fate.
Emerging hotspots and future directions of UME regulation in IBD
Building on the expanding mechanistic understanding of UME function in intestinal inflammation, efforts are now increasingly directed toward developing therapeutic and technological strategies to modulate UME activity in IBD (Figure 6).
Future perspectives of UMEs in IBD. Schematic summary of emerging research directions and therapeutic strategies targeting ubiquitin-modifying enzymes (UMEs) in inflammatory bowel disease, including small-molecule inhibitors, PROTAC technology, ncRNA-mediated regulation, natural compounds, multi-omics approaches, and clinical translational potential. Created in BioRender. Qian, C. (2026) https://BioRender.com/7t7alxc.
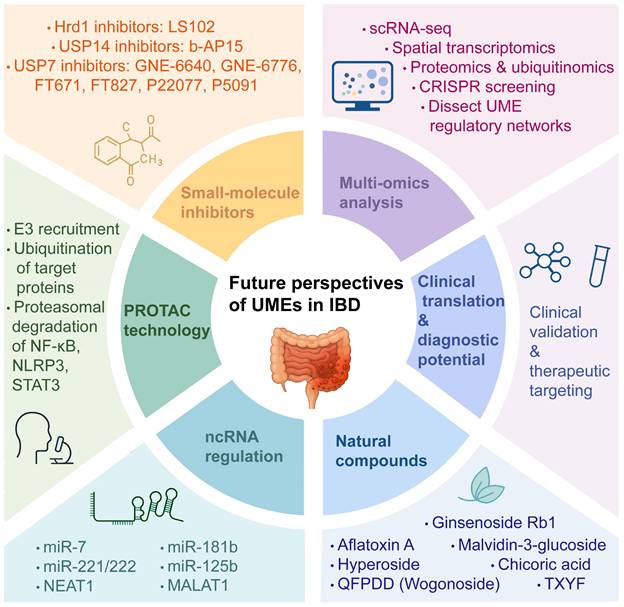
Development of small-molecule modulators targeting UMEs
With the increasing recognition of UMEs as pivotal regulators in chronic inflammatory diseases, small-molecule inhibitors targeting DUBs and E3 ligases have become a major research focus. Among them, USP7 and USP14, which regulate NF-κB signaling, inflammatory gene expression, and epithelial barrier homeostasis, have emerged as promising therapeutic targets in IBD.
Structural and functional diversity of USP7 small-molecule inhibitors
The therapeutic targeting of USP7 has evolved from early non-specific covalent inhibitors to highly selective allosteric modulators, driven by a deeper understanding of its complex structural dynamics [129, 130]. USP7 transitions between an inactive (apo) state and an active (ubiquitin-bound) state through significant rearrangements of its catalytic triad (Cys223, His464, and Asp481) [129].
Recent studies have identified small molecules that exploit USP7 conformational dynamics to achieve selective inhibition. GNE-6640 and GNE-6776 act as non-covalent inhibitors that bind near the catalytic cleft, induce rearrangement of the switching loop, and prevent ubiquitin engagement, thereby suppressing USP7 activity in cancer models [130]. In contrast, FT671 and FT827 selectively stabilize the apo conformation of USP7, block catalytic triad realignment, and promote MDM2 degradation, leading to restoration of p53 tumor-suppressive signaling [129]. Beyond oncology, USP7 has also emerged as a critical regulator of inflammatory signaling and redox homeostasis. P22077, an early-generation dual inhibitor of USP7 and USP47, exerts anti-inflammatory effects by targeting multiple signaling pathways. Mechanistically, P22077 promotes K48-linked ubiquitination and degradation of TRAF6, thereby attenuating LPS-induced TLR4/NF-κB activation [131]. In addition, USP7 inhibition by P22077 suppresses NLRP3 inflammasome activation by preventing ASC oligomerization and speck formation in a transcription-independent manner [132].
In the specific context of IBD, USP7 plays a deleterious role by modulating the intestinal redox state. Recent evidence highlights that USP7 stabilizes AMBRA1; the accumulated AMBRA1 then antagonizes DUB3-mediated deubiquitination of NRF2, leading to NRF2 degradation and increased oxidative stress in IECs. Consequently, inhibition of USP7 by P5091 or P22077 restores the NRF2-driven antioxidant response and alleviates mucosal inflammation in experimental colitis models [102, 133]. These findings suggest that USP7 inhibitors do not merely suppress inflammation but also act as multifaceted regulators of epithelial barrier integrity and oxidative balance, making them promising candidates for IBD therapy.
Small-molecule inhibitors targeting proteasome-associated USP14
USP14 is a major deubiquitinating enzyme associated with the 19S regulatory particle of the 26S proteasome and plays an essential role in fine-tuning proteasomal protein degradation. Unlike most DUBs, inhibition of USP14 directly interferes with proteasomal substrate processing, making it a potential therapeutic target in diseases characterized by proteostasis imbalance and inflammatory dysregulation.
b-AP15 is a potent small-molecule inhibitor targeting both USP14 and UCHL5. Its inhibition leads to rapid accumulation of high-molecular-weight polyubiquitinated proteins, thereby inducing pronounced proteotoxic stress [134]. In contrast to classical proteasome inhibitors, b-AP15-induced apoptosis is closely associated with enhanced oxidative stress and ROS generation, accompanied by mitochondrial membrane potential loss and structural damage, ultimately driving programmed cell death [135]. Beyond its pro-apoptotic effects in cancer models, b-AP15 also exhibits anti-inflammatory activity. USP14 inhibition attenuates LPS-induced inflammatory responses by suppressing ERK1/2 and JNK signaling and limiting NF-κB nuclear translocation [136]. By simultaneously modulating proteostasis and inflammatory signaling networks, targeting USP14 represents a multifaceted therapeutic strategy for inflammatory disorders.
Other small-molecule inhibitors targeting UMEs
Beyond the USP family, pharmacological modulation of other UMEs, particularly E3 ligases involved in ER stress and epithelial proteostasis, has also been explored. Among these, Hrd1 has attracted attention due to its critical role in regulating unfolded protein response signaling and maintaining epithelial stress tolerance. Experimental inhibition of Hrd1 using LS102 markedly exacerbates colitis severity in murine models, highlighting its protective function in limiting ER stress-driven epithelial injury and inflammatory amplification [62].
Collectively, these studies demonstrate that small-molecule modulation of UMEs can influence intestinal inflammation through multiple mechanisms, including inflammatory signal transduction, redox homeostasis, proteostasis regulation, and epithelial stress adaptation. While USP7 and USP14 inhibitors primarily act by reshaping inflammatory and proteotoxic signaling networks, targeting stress-responsive E3 ligases, such as Hrd1, underscores the importance of preserving epithelial homeostatic capacity. Together, these findings underscore both the therapeutic potential and the inherent complexity of UME-targeted strategies in IBD. Rational drug design will therefore require careful consideration of target selectivity, cell type specificity, and tissue-context dependence to balance anti-inflammatory efficacy with the maintenance of intestinal barrier integrity.
Natural compounds in the regulation of UMEs
Natural products are gaining attention as complementary regulators of UMEs. These bioactive compounds can influence UME expression or activity, thereby modulating intestinal inflammation, oxidative stress, and mitophagy.
For instance, in humanized colitis models, DSS or TNBS challenge significantly downregulated Hrd1 expression while upregulating ER stress markers such as GRP78, PERK, CHOP, and caspase-12. Treatment with ginsenoside Rb1 reversed these changes, suggesting a role in alleviating ER stress by modulating Hrd1 [62]. Similarly, aflatoxin A has been reported to promote the progression of chronic colitis and colorectal cancer by targeting the RINCK signaling pathway [137], underscoring the involvement of UMEs in IBD-associated tumorigenesis. Moreover, the traditional Chinese formula Tongxie Yaofang (TXYF) suppressed tumor formation in a CAC mouse model. TXYF not only inhibited the proliferation of LPS-stimulated epithelial and colorectal cancer cells but also blocked epithelial-mesenchymal transition (EMT) by activating PINK1/Parkin-dependent mitophagy, thereby exerting both anti-inflammatory and antitumor effects [138].
Among single compounds, Hyperoside was found to suppress MKRN1 expression, thereby enhancing ubiquitin-mediated degradation of its substrate, PPARγ, ultimately ameliorating DSS-induced colitis in mice [139]. Licorice extract has been shown to activate the Nrf2/PINK1 pathway, promoting mitophagy and reducing inflammatory cytokine production and oxidative stress in UC models [140]. Another study demonstrated that the blueberry anthocyanin malvidin-3-glucoside (MG) upregulated HACE1, restored gut microbial homeostasis, and increased the abundance of beneficial bacteria such as Bifidobacteria, conferring protection in colitis models [141]. Likewise, Qingfei Paidu Decoction (QFPDD) and its bioactive component wogonoside suppressed USP14 expression while promoting ATF2 degradation, resulting in reduced IL-6 and TNF-α production, enhanced IL-10 expression, and alleviation of intestinal inflammation [142]. Furthermore, Chicoric acid was shown to ameliorate DSS-induced colitis by targeting the USP9X/IGF2BP2 axis [143].
Regulation by long non-coding RNAs or microRNAs
In addition to classical regulation at the protein level, increasing evidence indicates that UMEs are also subject to fine-tuned control by non-coding RNA (ncRNA) networks [144]. Specific microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) can modulate the transcription, mRNA stability, or translational efficiency of UME-encoding genes, thereby indirectly influencing downstream signaling functions. Such regulatory layers play important roles in the onset and maintenance of intestinal inflammation [145, 146].
For instance, miR-7 suppresses RNF183 expression and alleviates DSS-induced colitis [147]. Conversely, miR-221/222 enhances NF-κB and STAT3 signaling activity through dual mechanisms: directly stabilizing RelA mRNA and indirectly repressing PDLIM2 (an E3 ligase that promotes the degradation of RelA and STAT3), thereby amplifying proinflammatory signals. Targeting miR-221/222 efficiently disrupts this pathogenic loop and inhibits colorectal cancer cell proliferation [148]. Furthermore, several miRNAs (e.g., miR-125b, miR-181b) and lncRNAs (e.g., NEAT1, MALAT1) have been experimentally validated to regulate key UMEs (such as A20, CYLD, and TRAF6), thereby modulating NF-κB signaling, cell survival, and immune homeostasis [149-153].
Although research on ncRNA-mediated regulation of UMEs in the context of IBD is still in its infancy, these findings highlight ncRNAs as upstream regulators of the UME network. Together, these findings expand our mechanistic understanding of UME regulation. Future studies integrating multi-cellular transcriptome profiling and ncRNA-UME interaction networks may help construct a multi-layered regulatory model, ultimately providing a more comprehensive explanation of UME functions in IBD.
Emerging PROTAC technology for targeting UMEs
In recent years, proteolysis-targeting chimera (PROTAC) technology has attracted considerable attention as a promising strategy for small-molecule drug development due to its ability to induce selective protein degradation [154]. Unlike conventional inhibitors that merely block enzymatic activity, PROTAC recruits an E3 ligase to ubiquitinate the target protein, thereby directing it toward proteasomal degradation. This provides a novel avenue for targeting “undruggable” proteins [155]. Although direct studies on UMEs in the context of IBD are still lacking, the successful application of PROTAC in oncology, autoimmunity, and other disease areas provides a strong theoretical basis for its potential application in IBD.
Of particular note, many UME-regulated proteins are highly stable and thus challenging to inhibit directly (e.g., certain DUBs or catalytically inactive E3 ligases), making them attractive PROTAC targets [156]. Future design strategies may consider developing PROTAC molecules against key IBD-related inflammatory mediators (such as NF-κB subunits, NLRP3, or STAT3). By harnessing E3 ligase-mediated selective degradation, such approaches could achieve precise regulation of inflammatory pathways. Furthermore, combining PROTAC platforms with tissue-specific delivery systems or biomaterial-based organ targeting strategies may further enhance their translational potential and therapeutic efficacy in IBD.
In summary, although PROTAC-based approaches are still in the early stages of research on intestinal inflammation, their mechanistic foundation in ubiquitin signaling highlights the promise of PROTAC as a powerful tool for modulating UMEs. This strategy may open new avenues for therapeutic intervention in IBD pathogenesis.
Multi-omics approaches for dissecting UME regulatory networks
The rapid development of multi-omics technologies has provided new perspectives for elucidating the regulatory roles of UMEs in IBD. Single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics have revealed cell-type- and tissue-specific expression patterns of UMEs, enabling precise localization of their activity in diseased tissues [157-159]. Integration of proteomic, ubiquitinomic, and transcriptomic data will facilitate the construction of comprehensive molecular regulatory networks of UMEs in intestinal immune inflammation, barrier dysfunction, and cell death. In addition, high-throughput CRISPR/Cas9 functional screens offer the opportunity to systematically identify essential UMEs and define their cell type- or pathway-specific roles. Together, these approaches will accelerate mechanistic dissection and uncover new therapeutic targets in IBD [160-162].
Clinical translation and diagnostic potential
Current evidence supporting the involvement of UMEs in IBD is primarily derived from heterogeneous human datasets, including bulk transcriptomic profiling, protein-level measurements, peripheral blood biomarker analyses, and genome-wide association studies (GWAS) [163, 164]. As summarized in the “Alteration in patients” category, many UMEs exhibit disease-associated expression changes in tissues from patients with UC, CD, or CRC (Figure 7, Tables 3-5). For example, UBC9, TRIM59, and USP13 are downregulated in patients with UC and CD and exhibit significant correlations with disease activity, indicating that alterations in UME expression may serve as indicators of disease progression or predictors of therapeutic response [25, 42, 71]. However, it should be emphasized that most patient-derived data reflect correlative associations rather than direct causal relationships. In contrast, only a subset of UMEs has been functionally validated in disease-relevant experimental systems, particularly inflammation-driven models such as AOM/DSS-induced CAC or genetically engineered colitis models. Therefore, caution is warranted when inferring mechanistic roles or therapeutic target potential based solely on observational human data. Future studies should prioritize the systematic validation of UMEs as biomarkers and companion diagnostics, alongside mechanistic investigations to establish their direct causal contributions to epithelial dysfunction, immune dysregulation, and inflammatory disease progression. Collectively, integrating multi-layer human datasets with functional validation in inflammation-driven models will be essential for refining the translational relevance of UMEs and prioritizing high-confidence targets for precision intervention in IBD and related inflammatory pathologies.
Representative UME expression alterations reported in heterogeneous patient cohorts with UC, CD, and CRC. Schematic summary of ubiquitin-modifying enzymes (UMEs) that are differentially expressed in ulcerative colitis (UC), Crohn's disease (CD), and colorectal cancer (CRC) based on reported transcriptomic and tissue-level expression analyses. Blue boxes indicate UMEs that are downregulated, whereas red boxes indicate UMEs that are upregulated in the indicated disease conditions. Enzymes altered in both UC and CD are shown separately from those selectively dysregulated in individual disease subtypes. The CRC-associated panel summarizes UMEs with consistent expression changes during colorectal tumorigenesis. Created in BioRender. Qian, C. (2026) https://BioRender.com/o0txak6.
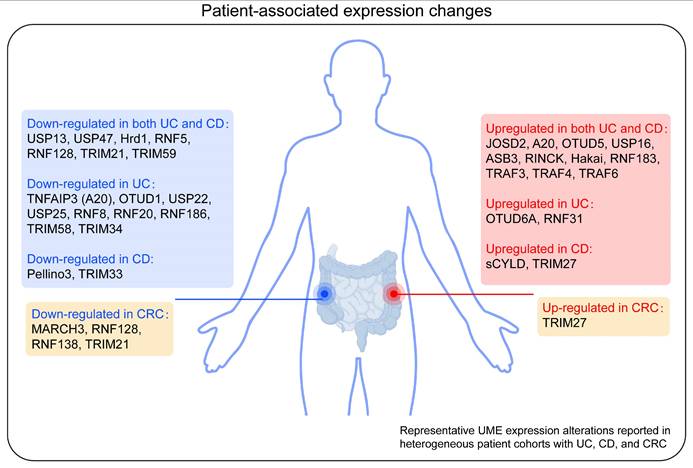
Conclusion, limitations, and future perspectives
By functioning as central regulators of protein homeostasis, UMEs play fundamental roles in controlling cell fate decisions during intestinal inflammation. By coordinating programmed cell death, stress adaptation, and survival signaling across epithelial and immune compartments, UMEs critically shape tissue homeostasis and inflammatory outcomes in IBD. This review has synthesized the current understanding of the expression dynamics and functional properties of E2 enzymes, E3 ligases, and DUBs, highlighting how ubiquitin-dependent mechanisms integrate cell-type-specific signaling networks under inflammatory stress.
Despite substantial progress, major conceptual and technical challenges remain in understanding UME-mediated regulation of intestinal inflammation. Key unresolved questions include the substrate specificity of individual UMEs, the functional selectivity of distinct ubiquitin chain architectures, tissue- and cell-type-dependent heterogeneity, and crosstalk with transcriptional and metabolic programs. In addition, the roles of UMEs in non-epithelial stromal populations, such as fibroblasts and endothelial cells, remain largely unexplored, particularly in the context of inflammation-associated tissue remodeling and disease complications. Limitations of current experimental models should also be considered. DSS- and TNBS-induced colitis models capture specific inflammatory features but do not fully recapitulate the chronic relapsing course, genetic diversity, and clinical heterogeneity of human IBD. DSS models primarily reflect epithelial injury and innate immune activation, whereas TNBS models emphasize hapten-driven, T cell-mediated inflammation. Likewise, AOM/DSS models preferentially represent colitis-associated tumorigenesis rather than sporadic colorectal cancer driven by oncogenic mutations. These limitations underscore the need for cautious translational interpretation and complementary validation across experimental systems.
Importantly, accumulating evidence indicates that UME function is highly stage-dependent along the trajectory of intestinal inflammation. During disease initiation, UMEs mainly shape epithelial barrier integrity, early innate immune activation, and acute epithelial cell death programs. In chronic inflammation, long-term regulation of inflammatory signaling, stress adaptation, and immune cell polarization contributes to disease persistence and tissue remodeling. In contrast, during CAC progression, dysregulation of epithelial survival pathways, genomic stability, and microenvironmental signaling becomes the dominant determinant of malignant progression. As summarized in Figure 4, many E3 ligases and DUBs exhibit context-dependent and bidirectional roles across epithelial, innate immune, adaptive immune, and tissue-level outcome layers. This stage-resolved perspective provides a conceptual framework for reconciling divergent experimental observations and highlights the importance of disease-stage stratification when interpreting UME function. From a translational standpoint, these findings suggest that therapeutic strategies targeting UMEs may require stage-stratified and context-adapted modulation rather than uniform inhibition or activation. For example, transient suppression of pro-inflammatory UMEs may be beneficial during acute inflammation, whereas preservation of epithelial-protective and stress-adaptive UMEs may be critical during chronic disease maintenance and CAC prevention.
Looking forward, integrating multi-omics profiling, spatial transcriptomics, and single-cell technologies will enable systematic mapping of ubiquitin-regulated cell-fate networks within complex inflammatory microenvironments. Combined with emerging chemical biology approaches, including small-molecule modulators and PROTAC-based strategies, these advances will facilitate mechanistic insight into cell death regulation and stress-responsive adaptation. Together, such efforts are anticipated to provide a robust conceptual framework for prioritizing high-confidence therapeutic targets and advancing precision, mechanism-driven interventions for inflammatory diseases.
Abbreviations
AOM: azoxymethane; ASC: apoptosis-associated speck-like protein containing a CARD; ATP: adenosine triphosphate; CAC: colitis-associated cancer; CD: Crohn's disease; CD4⁺: cluster of differentiation 4-positive; CHOP: C/EBP homologous protein; CLRs: C-type lectin receptors; CRC: colorectal cancer; CRISPR: clustered regularly interspaced short palindromic repeats; DCs: dendritic cells; DSS: dextran sulfate sodium; DUBs: deubiquitinating enzymes; E1: ubiquitin-activating enzyme; E2: ubiquitin-conjugating enzyme; E3: ubiquitin ligase; EMT: epithelial-mesenchymal transition; ER: endoplasmic reticulum; ERK1/2: extracellular signal-regulated kinase 1/2; GRP78: glucose-regulated protein 78; GWAS: genome-wide association studies; IBD: inflammatory bowel disease; IECs: intestinal epithelial cells; IGF2BP2: insulin-like growth factor 2 mRNA-binding protein 2; IKKβ: inhibitor of nuclear factor kappa-B kinase subunit beta; IL: interleukin; IRAKs: interleukin-1 receptor-associated kinases; IRF: interferon regulatory factor; JAMM: JAB1/MPN/Mov34 metalloenzyme; JNK: c-Jun N-terminal kinase; KDM5B: lysine demethylase 5B; KEAP1: Kelch-like ECH-associated protein 1; lncRNAs: long non-coding RNAs; LPS: lipopolysaccharide; MAPK: mitogen-activated protein kinase; miRNAs: microRNAs; MJD: Machado-Joseph disease; mRNA: messenger RNA; MyD88: myeloid differentiation primary response 88; ncRNA: non-coding RNA; NF-κB: nuclear factor kappa-B; NLRP3: NOD-like receptor family pyrin domain containing 3; NLRP6: NOD-like receptor family pyrin domain containing 6; NOD2: nucleotide-binding oligomerization domain-containing protein 2; NRF2: nuclear factor erythroid 2-related factor 2; OTU: ovarian tumor; PERK: protein kinase RNA-like endoplasmic reticulum kinase; PINK1: PTEN-induced kinase 1; PPARγ: peroxisome proliferator-activated receptor gamma; PROTAC: proteolysis-targeting chimera; PRRs: pattern recognition receptors; RelA: RELA proto-oncogene, NF-κB subunit; RIP2: receptor-interacting serine/threonine kinase 2; RIPK1: receptor-interacting protein kinase 1; ROS: reactive oxygen species; scRNA-seq: single-cell RNA sequencing; STAT3: signal transducer and activator of transcription 3; TAB: TAK1-binding protein; TAK1: transforming growth factor-beta-activated kinase 1; TGF-β: transforming growth factor-beta; Th1: T helper 1; Th17: T helper 17; TLR4: Toll-like receptor 4; TLRs: Toll-like receptors; TNBS: 2,4,6-trinitrobenzene sulfonic acid; TNF-α: tumor necrosis factor-alpha; TNFR1: tumor necrosis factor receptor 1; TNFRs: tumor necrosis factor receptors; Ub: ubiquitin; UBC9: ubiquitin-conjugating enzyme 9; UC: ulcerative colitis; UCHL5: ubiquitin C-terminal hydrolase L5; UMEs: ubiquitin-modifying enzymes; UPR: unfolded protein response; USP: ubiquitin-specific protease; VDR: vitamin D receptor.
Acknowledgements
Financial support was provided by the National Natural Science Foundation of China (82361138563 to Y.W.), the Innovation Team of Hangzhou City (TD2024002 to Y.W.), and Interdisciplinary Research Project of Hangzhou Normal University (2024JCXK06 to Y.W.).
AI usage statement
During the preparation of this manuscript, the author(s) used ChatGPT solely to improve the clarity and fluency of the English writing. After using this tool, the author(s) carefully reviewed and edited the text as needed and take full responsibility for the content of the manuscript.
Contributions
C.Q., F.N., C.Z., and Y.W. conceived and designed the review. C.Q. and Y.X. drafted the manuscript and prepared the tables and figures. C.Q., B.S., Y.X., and Y.W. participated in the literature search. C.Q., F.N., J.S., B.S., C.Z., and Y.X. contributed to the revision of the manuscript. All authors have read and approved the article.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kobayashi T, Siegmund B, Le Berre C, Wei SC, Ferrante M, Shen B. et al. Ulcerative colitis. Nature reviews Disease primers. 2020;6:74
2. Gajendran M, Loganathan P, Jimenez G, Catinella AP, Ng N, Umapathy C. et al. A comprehensive review and update on ulcerative colitis. Dis Mon. 2019;65:100851
3. Wan J, Zhou J, Wang Z, Liu D, Zhang H, Xie S. et al. Epidemiology, pathogenesis, diagnosis, and treatment of inflammatory bowel disease: Insights from the past two years. Chin Med J (Engl). 2025;138:763-76
4. Park JH, Peyrin-Biroulet L, Eisenhut M, Shin JI. IBD immunopathogenesis: A comprehensive review of inflammatory molecules. Autoimmun Rev. 2017;16:416-26
5. de Souza HS, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol. 2016;13:13-27
6. Neurath MF. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat Immunol. 2019;20:970-9
7. Jin X, You L, Qiao J, Han W, Pan H. Autophagy in colitis-associated colon cancer: exploring its potential role in reducing initiation and preventing IBD-Related CAC development. Autophagy. 2024;20:242-58
8. Sharma D, Kanneganti TD. Inflammatory cell death in intestinal pathologies. Immunol Rev. 2017;280:57-73
9. Wu J, Xu X, Duan J, Chai Y, Song J, Gong D. et al. EFHD2 suppresses intestinal inflammation by blocking intestinal epithelial cell TNFR1 internalization and cell death. Nat Commun. 2024;15:1282
10. Garcia-Hernandez V, Quiros M, Nusrat A. Intestinal epithelial claudins: expression and regulation in homeostasis and inflammation. Ann N Y Acad Sci. 2017;1397:66-79
11. Bain CC, Mowat AM. Macrophages in intestinal homeostasis and inflammation. Immunol Rev. 2014;260:102-17
12. Zhang M, Li X, Zhang Q, Yang J, Liu G. Roles of macrophages on ulcerative colitis and colitis-associated colorectal cancer. Front Immunol. 2023;14:1103617
13. Saez A, Herrero-Fernandez B, Gomez-Bris R, Sanchez-Martinez H, Gonzalez-Granado JM. Pathophysiology of Inflammatory Bowel Disease: Innate Immune System. Int J Mol Sci. 2023 24
14. Dikiy S, Ghelani AP, Levine AG, Martis S, Giovanelli P, Wang ZM. et al. Terminal differentiation and persistence of effector regulatory T cells essential for preventing intestinal inflammation. Nat Immunol. 2025;26:444-58
15. Thomson CA, Nibbs RJ, McCoy KD, Mowat AM. Immunological roles of intestinal mesenchymal cells. Immunology. 2020;160:313-24
16. Rogler G. Chronic ulcerative colitis and colorectal cancer. Cancer Lett. 2014;345:235-41
17. Shah SC, Itzkowitz SH. Colorectal Cancer in Inflammatory Bowel Disease: Mechanisms and Management. Gastroenterology. 2022;162:715-30 e3
18. Lill JR, Wertz IE. Toward understanding ubiquitin-modifying enzymes: from pharmacological targeting to proteomics. Trends Pharmacol Sci. 2014;35:187-207
19. Ruan J, Schluter D, Naumann M, Waisman A, Wang X. Ubiquitin-modifying enzymes as regulators of colitis. Trends Mol Med. 2022;28:304-18
20. Chen R, Pang X, Li L, Zeng Z, Chen M, Zhang S. Ubiquitin-specific proteases in inflammatory bowel disease-related signalling pathway regulation. Cell Death Dis. 2022;13:139
21. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY. et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119-24
22. Doms S, Fokt H, Ruhlemann MC, Chung CJ, Kuenstner A, Ibrahim SM. et al. Key features of the genetic architecture and evolution of host-microbe interactions revealed by high-resolution genetic mapping of the mucosa-associated gut microbiome in hybrid mice. Elife. 2022 11
23. Zhou L, Liu T, Huang B, Luo M, Chen Z, Zhao Z. et al. Excessive deubiquitination of NLRP3-R779C variant contributes to very-early-onset inflammatory bowel disease development. J Allergy Clin Immunol. 2021;147:267-79
24. Brant SR, Okou DT, Simpson CL, Cutler DJ, Haritunians T, Bradfield JP. et al. Genome-Wide Association Study Identifies African-Specific Susceptibility Loci in African Americans With Inflammatory Bowel Disease. Gastroenterology. 2017;152:206-17 e2
25. Qian C, Hu C, Xu Y, Xu W, Wang Z, Gan W. et al. Intestinal Epithelial-Derived USP13 Alleviates Colonic Inflammation by Suppressing GRP78-mediated Endoplasmic Reticulum Stress. Adv Sci (Weinh). 2025;12:e00741
26. Zhao B, Tsai YC, Jin B, Wang B, Wang Y, Zhou H. et al. Protein Engineering in the Ubiquitin System: Tools for Discovery and Beyond. Pharmacological reviews. 2020;72:380-413
27. Buetow L, Huang DT. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat Rev Mol Cell Biol. 2016;17:626-42
28. Lee JS, Kim HY, Kwon YT, Ji CH, Lee SJ, Kim SB. The Ubiquitin Code in Disease Pathogenesis and Progression: Composition, Characteristics and its Potential as a Therapeutic Target. Discov Med. 2025;37:203-21
29. Rahman S, Wolberger C. Breaking the K48-chain: linking ubiquitin beyond protein degradation. Nat Struct Mol Biol. 2024;31:216-8
30. Baur R, Rape M. Getting Close: Insight into the Structure and Function of K11/K48-Branched Ubiquitin Chains. Structure. 2020;28:1-3
31. Breeze E. Ubiquitous Ubiquitin: The K63 Ubiquitinome. Plant Cell. 2020;32:8-9
32. Livneh I, Kravtsova-Ivantsiv Y, Braten O, Kwon YT, Ciechanover A. Monoubiquitination joins polyubiquitination as an esteemed proteasomal targeting signal. Bioessays. 2017 39
33. Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature. 2016;535:75-84
34. Parashar S, Kaushik A, Ambasta RK, Kumar P. E2 conjugating enzymes: A silent but crucial player in ubiquitin biology. Ageing Res Rev. 2025;108:102740
35. Zhang H, Hu H, Greeley N, Jin J, Matthews AJ, Ohashi E. et al. STAT3 restrains RANK- and TLR4-mediated signalling by suppressing expression of the E2 ubiquitin-conjugating enzyme Ubc13. Nat Commun. 2014;5:5798
36. Eldridge MJG, Sanchez-Garrido J, Hoben GF, Goddard PJ, Shenoy AR. The Atypical Ubiquitin E2 Conjugase UBE2L3 Is an Indirect Caspase-1 Target and Controls IL-1beta Secretion by Inflammasomes. Cell Rep. 2017;18:1285-97
37. Kathania M, Zeng M, Yadav VN, Moghaddam SJ, Yang B, Venuprasad K. Ndfip1 regulates itch ligase activity and airway inflammation via UbcH7. J Immunol. 2015;194:2160-7
38. Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203-29
39. Hetzenecker AM, Seidl MC, Kosovac K, Herfarth H, Kellermeier S, Obermeier F. et al. Downregulation of the ubiquitin-proteasome system in normal colonic macrophages and reinduction in inflammatory bowel disease. Digestion. 2012;86:34-47
40. Wang S, Pu J, Li X, Yan Z, Li C, Zheng Y. et al. UBE2W Improves the Experimental Colitis by Inhibiting the NF-kappaB Signaling Pathway. Dig Dis Sci. 2022;67:5529-39
41. Kumar R, Singh AK, Starokadomskyy P, Luo W, Theiss AL, Burstein E. et al. Cutting Edge: Hypoxia-Induced Ubc9 Promoter Hypermethylation Regulates IL-17 Expression in Ulcerative Colitis. J Immunol. 2021;206:936-40
42. Mustfa SA, Singh M, Suhail A, Mohapatra G, Verma S, Chakravorty D. et al. SUMOylation pathway alteration coupled with downregulation of SUMO E2 enzyme at mucosal epithelium modulates inflammation in inflammatory bowel disease. Open Biol. 2017 7
43. Zhang J, Chen L, Xu Q, Zou Y, Sun F, Zhou Q. et al. Ubc9 regulates the expression of MHC II in dendritic cells to enhance DSS-induced colitis by mediating RBPJ SUMOylation. Cell Death Dis. 2023;14:737
44. Smit JJ, Sixma TK. RBR E3-ligases at work. EMBO Rep. 2014;15:142-54
45. Cleynen I, Vazeille E, Artieda M, Verspaget HW, Szczypiorska M, Bringer MA. et al. Genetic and microbial factors modulating the ubiquitin proteasome system in inflammatory bowel disease. Gut. 2014;63:1265-74
46. Tortola L, Nitsch R, Bertrand MJM, Kogler M, Redouane Y, Kozieradzki I. et al. The Tumor Suppressor Hace1 Is a Critical Regulator of TNFR1-Mediated Cell Fate. Cell Rep. 2016;15:1481-92
47. Liang J, Wang N, Yao Y, Wang Y, An X, Wang H. et al. NEDD4L mediates intestinal epithelial cell ferroptosis to restrict inflammatory bowel diseases and colorectal tumorigenesis. J Clin Invest. 2024 135
48. Fujimoto K, Kinoshita M, Tanaka H, Okuzaki D, Shimada Y, Kayama H. et al. Regulation of intestinal homeostasis by the ulcerative colitis-associated gene RNF186. Mucosal Immunol. 2017;10:446-59
49. Zhang H, Cui Z, Cheng D, Du Y, Guo X, Gao R. et al. RNF186 regulates EFNB1 (ephrin B1)-EPHB2-induced autophagy in the colonic epithelial cells for the maintenance of intestinal homeostasis. Autophagy. 2021;17:3030-47
50. Zhao J, Cai B, Shao Z, Zhang L, Zheng Y, Ma C. et al. TRIM26 positively regulates the inflammatory immune response through K11-linked ubiquitination of TAB1. Cell Death Differ. 2021;28:3077-91
51. Song H, Liu B, Huai W, Yu Z, Wang W, Zhao J. et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat Commun. 2016;7:13727
52. Hwang S, Park J, Koo SY, Lee SY, Jo Y, Ryu D. et al. The ubiquitin ligase Pellino1 targets STAT3 to regulate macrophage-mediated inflammation and tumor development. Nat Commun. 2025;16:1256
53. Kim YM, Kim HY, Ha Thi HT, Kim J, Lee YJ, Kim SJ. et al. Pellino 3 promotes the colitis-associated colorectal cancer through suppression of IRF4-mediated negative regulation of TLR4 signalling. Mol Oncol. 2023;17:2380-95
54. Kathania M, Khare P, Zeng M, Cantarel B, Zhang H, Ueno H. et al. Itch inhibits IL-17-mediated colon inflammation and tumorigenesis by ROR-gammat ubiquitination. Nat Immunol. 2016;17:997-1004
55. Kathania M, Tsakem EL, Theiss AL, Venuprasad K. Gut Microbiota Contributes to Spontaneous Colitis in E3 Ligase Itch-Deficient Mice. J Immunol. 2020;204:2277-84
56. Fujita Y, Khateb A, Li Y, Tinoco R, Zhang T, Bar-Yoseph H. et al. Regulation of S100A8 Stability by RNF5 in Intestinal Epithelial Cells Determines Intestinal Inflammation and Severity of Colitis. Cell Rep. 2018;24:3296-311 e6
57. Shang J, Li L, Wang X, Pan H, Liu S, He R. et al. Disruption of Tumor Necrosis Factor Receptor-Associated Factor 5 Exacerbates Murine Experimental Colitis via Regulating T Helper Cell-Mediated Inflammation. Mediators Inflamm. 2016;2016:9453745
58. Zhou G, Wu W, Yu L, Yu T, Yang W, Wang P. et al. Tripartite motif-containing (TRIM) 21 negatively regulates intestinal mucosal inflammation through inhibiting T(H)1/T(H)17 cell differentiation in patients with inflammatory bowel diseases. J Allergy Clin Immunol. 2018;142:1218-28 e12
59. Chen Z, Lin B, Yao X, Fang Y, Liu J, Song K. et al. OAS3 Deubiquitination Due to E3 Ligase TRIM21 Downregulation Promotes Epithelial Cell Apoptosis and Drives Sepsis-induced Acute Lung Injury. Int J Biol Sci. 2024;20:5594-607
60. Qi S, Li Y, Dai Z, Xiang M, Wang G, Wang L. et al. Uhrf1-Mediated Tnf-alpha Gene Methylation Controls Proinflammatory Macrophages in Experimental Colitis Resembling Inflammatory Bowel Disease. J Immunol. 2019;203:3045-53
61. Grabinger T, Bode KJ, Demgenski J, Seitz C, Delgado ME, Kostadinova F. et al. Inhibitor of Apoptosis Protein-1 Regulates Tumor Necrosis Factor-Mediated Destruction of Intestinal Epithelial Cells. Gastroenterology. 2017;152:867-79
62. Dong JY, Xia KJ, Liang W, Liu LL, Yang F, Fang XS. et al. Ginsenoside Rb1 alleviates colitis in mice via activation of endoplasmic reticulum-resident E3 ubiquitin ligase Hrd1 signaling pathway. Acta Pharmacol Sin. 2021;42:1461-71
63. Wu Y, Kimura Y, Okamoto T, Matsuhisa K, Asada R, Saito A. et al. Inflammatory bowel disease-associated ubiquitin ligase RNF183 promotes lysosomal degradation of DR5 and TRAIL-induced caspase activation. Sci Rep. 2019;9:20301
64. Ma Z, Wu J, Wu Y, Sun X, Rao Z, Sun N. et al. Parkin increases the risk of colitis by downregulation of VDR via autophagy-lysosome degradation. Int J Biol Sci. 2023;19:1633-44
65. Jiang H, Xie Y, Hu Z, Lu J, Zhang J, Li H. et al. VANGL2 alleviates inflammatory bowel disease by recruiting the ubiquitin ligase MARCH8 to limit NLRP3 inflammasome activation through OPTN-mediated selective autophagy. PLoS Biol. 2025;23:e3002961
66. Zhu Y, Shi Y, Ke X, Xuan L, Ma Z. RNF8 induces autophagy and reduces inflammation by promoting AKT degradation via ubiquitination in ulcerative colitis mice. J Biochem. 2020;168:445-53
67. Ran X, Li Y, Ren Y, Chang W, Deng R, Wang H. et al. RNF128 deficiency in macrophages promotes colonic inflammation by suppressing the autophagic degradation of S100A8. Cell death & disease. 2025;16:20
68. Liu X, Yan C, Chang C, Meng F, Shen W, Wang S. et al. Ochratoxin A promotes chronic enteritis and early colorectal cancer progression by targeting Rinck signaling. Phytomedicine. 2024;122:155095
69. Tang CT, Liu ZD, Wang P, Zeng CY, Chen YX. Lipopolysaccharide-regulated RNF31/NRF2 axis in colonic epithelial cells mediates homeostasis of the intestinal barrier in ulcerative colitis. Cell Signal. 2024;124:111480
70. Roca-Lema D, Quiroga M, Khare V, Diaz-Diaz A, Barreiro-Alonso A, Rodriguez-Alonso A. et al. Role of the E3 ubiquitin-ligase Hakai in intestinal inflammation and cancer bowel disease. Sci Rep. 2022;12:17571
71. Liu B, Gao Y, Liu X, Lian Q, Li Y. Tripartite motif containing 59 mediates protective anti-oxidative effects in intestinal injury through Nrf2 signaling. Int Immunopharmacol. 2023;124:110896
72. Lin H, Feng L, Cui KS, Zeng LW, Gao D, Zhang LX. et al. The membrane-associated E3 ubiquitin ligase MARCH3 downregulates the IL-6 receptor and suppresses colitis-associated carcinogenesis. Cell Mol Immunol. 2021;18:2648-59
73. Zhou G, Wu H, Lin J, Lin R, Feng B, Liu Z. TRIM21 Is Decreased in Colitis-associated Cancer and Negatively Regulates Epithelial Carcinogenesis. Inflamm Bowel Dis. 2021;27:458-68
74. Zhang HX, Xu ZS, Lin H, Li M, Xia T, Cui K. et al. TRIM27 mediates STAT3 activation at retromer-positive structures to promote colitis and colitis-associated carcinogenesis. Nat Commun. 2018;9:3441
75. Lian Q, Yan S, Yin Q, Yan C, Zheng W, Gu W. et al. TRIM34 attenuates colon inflammation and tumorigenesis by sustaining barrier integrity. Cell Mol Immunol. 2021;18:350-62
76. Wang P, Tang CT, Li J, Huang X, Jin R, Yin F. et al. The E3 ubiquitin ligase RNF31 mediates the development of ulcerative colitis by regulating NLRP3 inflammasome activation. Int Immunopharmacol. 2023;125:111194
77. Petit V, Parcelier A, Mathe C, Barroca V, Torres C, Lewandowski D. et al. TRIM33 deficiency in monocytes and macrophages impairs resolution of colonic inflammation. EBioMedicine. 2019;44:60-70
78. Duan JL, He HQ, Yu Y, Liu T, Ma SJ, Li F. et al. E3 ligase c-Cbl regulates intestinal inflammation through suppressing fungi-induced noncanonical NF-kappaB activation. Sci Adv. 2021 7
79. Jin J, Xiao Y, Hu H, Zou Q, Li Y, Gao Y. et al. Proinflammatory TLR signalling is regulated by a TRAF2-dependent proteolysis mechanism in macrophages. Nat Commun. 2015;6:5930
80. Kosinsky RL, Chua RL, Qui M, Saul D, Mehlich D, Strobel P. et al. Loss of RNF40 Decreases NF-kappaB Activity in Colorectal Cancer Cells and Reduces Colitis Burden in Mice. J Crohns Colitis. 2019;13:362-73
81. Shen J, Qiao Y, Ran Z, Wang T. Different activation of TRAF4 and TRAF6 in inflammatory bowel disease. Mediators of inflammation. 2013;2013:647936
82. Chen M, Zhao Z, Meng Q, Liang P, Su Z, Wu Y. et al. TRIM14 Promotes Noncanonical NF-kappaB Activation by Modulating p100/p52 Stability via Selective Autophagy. Adv Sci (Weinh). 2020;7:1901261
83. Bertrand MJ, Doiron K, Labbe K, Korneluk RG, Barker PA, Saleh M. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity. 2009;30:789-801
84. Zhang J, Cao L, Gao A, Ren R, Yu L, Li Q. et al. E3 ligase RNF99 negatively regulates TLR-mediated inflammatory immune response via K48-linked ubiquitination of TAB2. Cell Death Differ. 2023;30:966-78
85. Lu Y, Huang R, Ying J, Li X, Jiao T, Guo L. et al. RING finger 138 deregulation distorts NF-small ka, CyrillicB signaling and facilities colitis switch to aggressive malignancy. Signal Transduct Target Ther. 2022;7:185
86. Zurek B, Schoultz I, Neerincx A, Napolitano LM, Birkner K, Bennek E. et al. TRIM27 negatively regulates NOD2 by ubiquitination and proteasomal degradation. PLoS One. 2012;7:e41255
87. Eyking A, Ferber F, Kohler S, Reis H, Cario E. TRIM58 Restrains Intestinal Mucosal Inflammation by Negatively Regulating TLR2 in Myeloid Cells. Journal of immunology (Baltimore, Md: 1950). 2019;203:1636-49
88. Sun X, Cui Y, Feng H, Liu H, Liu X. TGF-beta signaling controls Foxp3 methylation and T reg cell differentiation by modulating Uhrf1 activity. J Exp Med. 2019;216:2819-37
89. He TS, Cai K, Lai W, Yu J, Qing F, Shen A. et al. E3 ubiquitin ligase RNF128 attenuates colitis and colorectal tumorigenesis by triggering the degradation of IL-6 receptors. J Adv Res. 2024
90. Paul J, Singh AK, Kathania M, Elviche TL, Zeng M, Basrur V. et al. IL-17-driven intestinal fibrosis is inhibited by Itch-mediated ubiquitination of HIC-5. Mucosal Immunol. 2018;11:427-36
91. Wang P, Wu Y, Li Y, Zheng J, Tang J. A novel RING finger E3 ligase RNF186 regulate ER stress-mediated apoptosis through interaction with BNip1. Cell Signal. 2013;25:2320-33
92. Ranjan K, Hedl M, Sinha S, Zhang X, Abraham C. Ubiquitination of ATF6 by disease-associated RNF186 promotes the innate receptor-induced unfolded protein response. J Clin Invest. 2021 131
93. Lange SM, Armstrong LA, Kulathu Y. Deubiquitinases: From mechanisms to their inhibition by small molecules. Mol Cell. 2022;82:15-29
94. Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10:550-63
95. Harrigan JA, Jacq X, Martin NM, Jackson SP. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat Rev Drug Discov. 2018;17:57-78
96. Zou M, Zeng QS, Nie J, Yang JH, Luo ZY, Gan HT. The Role of E3 Ubiquitin Ligases and Deubiquitinases in Inflammatory Bowel Disease: Friend or Foe? Front Immunol. 2021;12:769167
97. Verboom L, Anderson CJ, Jans M, Petta I, Blancke G, Martens A. et al. OTULIN protects the intestinal epithelium from apoptosis during inflammation and infection. Cell death & disease. 2023;14:534
98. Khan OM, Carvalho J, Spencer-Dene B, Mitter R, Frith D, Snijders AP. et al. The deubiquitinase USP9X regulates FBW7 stability and suppresses colorectal cancer. J Clin Invest. 2018;128:1326-37
99. Kosinsky RL, Saul D, Ammer-Herrmenau C, Faubion WA, Neesse A, Johnsen SA. USP22 Suppresses SPARC Expression in Acute Colitis and Inflammation-Associated Colorectal Cancer. Cancers (Basel). 2021 13
100. Liu Z, Liu J, Wei Y, Li J, Zhang J, Yu R. et al. Ubiquitin-specific protease 25 ameliorates ulcerative colitis by regulating the degradation of phosphor-STAT3. Cell death & disease. 2025;16:5
101. Lei H, Yang L, Xu H, Wang Z, Li X, Liu M. et al. Ubiquitin-specific protease 47 regulates intestinal inflammation through deubiquitination of TRAF6 in epithelial cells. Sci China Life Sci. 2022;65:1624-35
102. Xu W, Hua Z, Wang Y, Tang W, Ge W, Chen Y. et al. Redox-Induced Stabilization of AMBRA1 by USP7 Promotes Intestinal Oxidative Stress and Colitis Through Antagonizing DUB3-Mediated NRF2 Deubiquitination. Adv Sci (Weinh). 2025;12:e2411320
103. Garcia-Carbonell R, Wong J, Kim JY, Close LA, Boland BS, Wong TL. et al. Elevated A20 promotes TNF-induced and RIPK1-dependent intestinal epithelial cell death. Proc Natl Acad Sci U S A. 2018;115:E9192-E200
104. Vereecke L, Sze M, Mc Guire C, Rogiers B, Chu Y, Schmidt-Supprian M. et al. Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor-induced toxicity and experimental colitis. The Journal of experimental medicine. 2010;207:1513-23
105. Mukherjee S, Kumar R, Tsakem Lenou E, Basrur V, Kontoyiannis DL, Ioakeimidis F. et al. Deubiquitination of NLRP6 inflammasome by Cyld critically regulates intestinal inflammation. Nat Immunol. 2020;21:626-35
106. Karatzas DN, Xanthopoulos K, Kotantaki P, Pseftogas A, Teliousis K, Hatzivassiliou EG. et al. Inactivation of CYLD in intestinal epithelial cells exacerbates colitis-associated colorectal carcinogenesis - a short report. Cell Oncol (Dordr). 2016;39:287-93
107. Hammer GE, Turer EE, Taylor KE, Fang CJ, Advincula R, Oshima S. et al. Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat Immunol. 2011;12:1184-93
108. Liu X, Fang Y, Huang M, Tu S, Zheng B, Yuan H. et al. Deubiquitinase JOSD2 alleviates colitis by inhibiting inflammation via deubiquitination of IMPDH2 in macrophages. Acta Pharm Sin B. 2025;15:1039-55
109. Zhang J, Stirling B, Temmerman ST, Ma CA, Fuss IJ, Derry JM. et al. Impaired regulation of NF-kappaB and increased susceptibility to colitis-associated tumorigenesis in CYLD-deficient mice. J Clin Invest. 2006;116:3042-9
110. Zhao Z, Su Z, Liang P, Liu D, Yang S, Wu Y. et al. USP38 Couples Histone Ubiquitination and Methylation via KDM5B to Resolve Inflammation. Adv Sci (Weinh). 2020;7:2002680
111. Kim JS, Kim HK, Lee J, Jang S, Cho E, Mun SJ. et al. Inhibition of CD82 improves colitis by increasing NLRP3 deubiquitination by BRCC3. Cell Mol Immunol. 2023;20:189-200
112. Dinallo V, Di Fusco D, Di Grazia A, Laudisi F, Troncone E, Di Maggio G. et al. The Deubiquitinating Enzyme OTUD5 Sustains Inflammatory Cytokine Response in Inflammatory Bowel Disease. J Crohns Colitis. 2022;16:122-32
113. Liu X, Fang Y, Lv X, Hu C, Chen G, Zhang L. et al. Deubiquitinase OTUD6A in macrophages promotes intestinal inflammation and colitis via deubiquitination of NLRP3. Cell Death Differ. 2023;30:1457-71
114. Yu JS, Huang T, Zhang Y, Mao XT, Huang LJ, Li YN. et al. Substrate-specific recognition of IKKs mediated by USP16 facilitates autoimmune inflammation. Sci Adv. 2021 7
115. Wang XM, Yang C, Zhao Y, Xu ZG, Yang W, Wang P. et al. The deubiquitinase USP25 supports colonic inflammation and bacterial infection and promotes colorectal cancer. Nat Cancer. 2020;1:811-25
116. van Loosdregt J, Fleskens V, Fu J, Brenkman AB, Bekker CP, Pals CE. et al. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity. 2013;39:259-71
117. Dufner A, Kisser A, Niendorf S, Basters A, Reissig S, Schonle A. et al. The ubiquitin-specific protease USP8 is critical for the development and homeostasis of T cells. Nat Immunol. 2015;16:950-60
118. Wang L, Cheng H, Wang X, Zhu F, Tian N, Xu Z. et al. Deubiquitination of aryl hydrocarbon receptor by USP21 negatively regulates T helper 17 cell differentiation. J Leukoc Biol. 2024 117
119. Le Menn G, Pikkarainen K, Mennerich D, Miroszewska D, Kietzmann T, Chen Z. USP28 protects development of inflammation in mouse intestine by regulating STAT5 phosphorylation and IL22 production in T lymphocytes. Front Immunol. 2024;15:1401949
120. Tang Y, Reissig S, Glasmacher E, Regen T, Wanke F, Nikolaev A. et al. Alternative Splice Forms of CYLD Mediate Ubiquitination of SMAD7 to Prevent TGFB Signaling and Promote Colitis. Gastroenterology. 2019;156:692-707 e7
121. Lee SH, Lee HR, Kwon JY, Jung K, Kim SY, Cho KH. et al. A20 ameliorates inflammatory bowel disease in mice via inhibiting NF-kappaB and STAT3 activation. Immunol Lett. 2018;198:44-51
122. Yang S, Wang B, Humphries F, Jackson R, Healy ME, Bergin R. et al. Pellino3 ubiquitinates RIP2 and mediates Nod2-induced signaling and protective effects in colitis. Nat Immunol. 2013;14:927-36
123. Cheng M, Xu B, Sun Y, Wang J, Lu Y, Shi C. et al. ASB3 expression aggravates inflammatory bowel disease by targeting TRAF6 protein stability and affecting the intestinal microbiota. mBio. 2024;15:e0204324
124. Cao Z, Conway KL, Heath RJ, Rush JS, Leshchiner ES, Ramirez-Ortiz ZG. et al. Ubiquitin Ligase TRIM62 Regulates CARD9-Mediated Anti-fungal Immunity and Intestinal Inflammation. Immunity. 2015;43:715-26
125. Wei J, Wei C, Wang M, Qiu X, Li Y, Yuan Y. et al. The GTPase-activating protein GIT2 protects against colitis by negatively regulating Toll-like receptor signaling. Proc Natl Acad Sci U S A. 2014;111:8883-8
126. Wu B, Qiang L, Zhang Y, Fu Y, Zhao M, Lei Z. et al. The deubiquitinase OTUD1 inhibits colonic inflammation by suppressing RIPK1-mediated NF-kappaB signaling. Cell Mol Immunol. 2022;19:276-89
127. Hou P, Jia P, Yang K, Li Z, Tian T, Lin Y. et al. An unconventional role of an ASB family protein in NF-kappaB activation and inflammatory response during microbial infection and colitis. Proc Natl Acad Sci U S A. 2021 118
128. Tarcic O, Pateras IS, Cooks T, Shema E, Kanterman J, Ashkenazi H. et al. RNF20 Links Histone H2B Ubiquitylation with Inflammation and Inflammation-Associated Cancer. Cell Rep. 2016;14:1462-76
129. Turnbull AP, Ioannidis S, Krajewski WW, Pinto-Fernandez A, Heride C, Martin ACL. et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature. 2017;550:481-6
130. Kategaya L, Di Lello P, Rouge L, Pastor R, Clark KR, Drummond J. et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature. 2017;550:534-8
131. Zhao XB, Ji FY, Li HR, Zhu HH, Zhao ZZ, Ling J. et al. P22077 inhibits LPS-induced inflammatory response by promoting K48-linked ubiquitination and degradation of TRAF6. Aging (Albany NY). 2020;12:10969-82
132. Palazon-Riquelme P, Worboys JD, Green J, Valera A, Martin-Sanchez F, Pellegrini C. et al. USP7 and USP47 deubiquitinases regulate NLRP3 inflammasome activation. EMBO Rep. 2018 19
133. Zeng D, Zhang W, Chen X, Ou G, Huang Y, Yu C. Inhibitory Effect of P22077 on Airway Inflammation in Rats with COPD and Its Mechanism. Int J Chron Obstruct Pulmon Dis. 2024;19:779-88
134. Zhang X, Pellegrini P, Saei AA, Hillert EK, Mazurkiewicz M, Olofsson MH. et al. The deubiquitinase inhibitor b-AP15 induces strong proteotoxic stress and mitochondrial damage. Biochem Pharmacol. 2018;156:291-301
135. Brnjic S, Mazurkiewicz M, Fryknas M, Sun C, Zhang X, Larsson R. et al. Induction of tumor cell apoptosis by a proteasome deubiquitinase inhibitor is associated with oxidative stress. Antioxid Redox Signal. 2014;21:2271-85
136. Zhang F, Xu R, Chai R, Xu Q, Liu M, Chen X. et al. Deubiquitinase Inhibitor b-AP15 Attenuated LPS-Induced Inflammation via Inhibiting ERK1/2, JNK, and NF-Kappa B. Front Mol Biosci. 2020;7:49
137. Zhang L, Chen J, Yang X, Shen C, Huang J, Zhang D. et al. Hepatic Zbtb18 (Zinc Finger and BTB Domain Containing 18) alleviates hepatic steatohepatitis via FXR (Farnesoid X Receptor). Signal Transduct Target Ther. 2024;9:20
138. Xu Z, Zhao G, Zhang L, Qiao C, Wang H, Wei H. et al. Tong-Xie-Yao-Fang induces mitophagy in colonic epithelial cells to inhibit colitis-associated colorectal cancer. J Ethnopharmacol. 2024;334:118541
139. Cheng C, Zhang W, Zhang C, Ji P, Wu X, Sha Z. et al. Hyperoside Ameliorates DSS-Induced Colitis through MKRN1-Mediated Regulation of PPARgamma Signaling and Th17/Treg Balance. J Agric Food Chem. 2021;69:15240-51
140. Kong J, Xiang Q, Shi G, Xu Z, Ma X, Wang Y. et al. Licorice protects against ulcerative colitis via the Nrf2/PINK1-mediated mitochondrial autophagy. Immun Inflamm Dis. 2023;11:e757
141. Liu F, Smith AD, Wang TTY, Pham Q, Cheung L, Yang H. et al. Biological pathways via which the anthocyanin malvidin alleviated the murine colitis induced by Citrobacter rodentium. Food Funct. 2023;14:1048-61
142. Xu X, Xia J, Zhao S, Wang Q, Ge G, Xu F. et al. Qing-Fei-Pai-Du decoction and wogonoside exert anti-inflammatory action through down-regulating USP14 to promote the degradation of activating transcription factor 2. FASEB J. 2021;35:e21870
143. Chen W, Shan Y, Wang M, Liang R, Sa R. Chicoric acid exerts therapeutic effects in DSS-induced ulcerative colitis by targeting the USP9X/IGF2BP2 axis. British journal of pharmacology. 2024
144. You JR, Wen ZJ, Tian JW, Lv XB, Li R, Li SP. et al. Crosstalk between ubiquitin ligases and ncRNAs drives cardiovascular disease progression. Front Immunol. 2024;15:1335519
145. Zhou J, Liu J, Gao Y, Shen L, Li S, Chen S. miRNA-Based Potential Biomarkers and New Molecular Insights in Ulcerative Colitis. Front Pharmacol. 2021;12:707776
146. Rankin CR, Theodorou E, Man Law IK, Rowe L, Kokkotou E, Pekow J. et al. Identification of novel mRNAs and lncRNAs associated with mouse experimental colitis and human inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2018;315:G722-G33
147. Yu Q, Zhang S, Chao K, Feng R, Wang H, Li M. et al. E3 Ubiquitin ligase RNF183 Is a Novel Regulator in Inflammatory Bowel Disease. J Crohns Colitis. 2016;10:713-25
148. Liu S, Sun X, Wang M, Hou Y, Zhan Y, Jiang Y. et al. A microRNA 221- and 222-mediated feedback loop maintains constitutive activation of NFkappaB and STAT3 in colorectal cancer cells. Gastroenterology. 2014;147:847-59 e11
149. Li LN, Xiao T, Yi HM, Zheng Z, Qu JQ, Huang W. et al. MiR-125b Increases Nasopharyngeal Carcinoma Radioresistance by Targeting A20/NF-kappaB Signaling Pathway. Mol Cancer Ther. 2017;16:2094-106
150. Zheng Z, Qu JQ, Yi HM, Ye X, Huang W, Xiao T. et al. MiR-125b regulates proliferation and apoptosis of nasopharyngeal carcinoma by targeting A20/NF-kappaB signaling pathway. Cell death & disease. 2017;8:e2855
151. Parisi C, Napoli G, Amadio S, Spalloni A, Apolloni S, Longone P. et al. MicroRNA-125b regulates microglia activation and motor neuron death in ALS. Cell Death Differ. 2016;23:531-41
152. Song L, Lin C, Gong H, Wang C, Liu L, Wu J. et al. miR-486 sustains NF-kappaB activity by disrupting multiple NF-kappaB-negative feedback loops. Cell Res. 2013;23:274-89
153. Du M, Yuan L, Tan X, Huang D, Wang X, Zheng Z. et al. The LPS-inducible lncRNA Mirt2 is a negative regulator of inflammation. Nat Commun. 2017;8:2049
154. Samarasinghe KTG, Crews CM. Targeted protein degradation: A promise for undruggable proteins. Cell Chem Biol. 2021;28:934-51
155. Xiong Y, Zhong Y, Yim H, Yang X, Park KS, Xie L. et al. Bridged Proteolysis Targeting Chimera (PROTAC) Enables Degradation of Undruggable Targets. J Am Chem Soc. 2022;144:22622-32
156. Osei-Amponsa V, Walters KJ. Proteasome substrate receptors and their therapeutic potential. Trends Biochem Sci. 2022;47:950-64
157. Doherty LM, Mills CE, Boswell SA, Liu X, Hoyt CT, Gyori B. et al. Integrating multi-omics data reveals function and therapeutic potential of deubiquitinating enzymes. Elife. 2022 11
158. Liu Y, Zhang X, Yu L, Cao L, Zhang J, Li Q. et al. E3 ubiquitin ligase RNF128 promotes Lys63-linked polyubiquitination on SRB1 in macrophages and aggravates atherosclerosis. Nat Commun. 2025;16:2185
159. Lu C, Liu H, Liu T, Sun S, Zheng Y, Ling T. et al. RIPK2 promotes colorectal cancer metastasis by protecting YAP degradation from ITCH-mediated ubiquitination. Cell death & disease. 2025;16:248
160. Cortez JT, Montauti E, Shifrut E, Gatchalian J, Zhang Y, Shaked O. et al. CRISPR screen in regulatory T cells reveals modulators of Foxp3. Nature. 2020;582:416-20
161. Chen W, Yang KB, Zhang YZ, Lin ZS, Chen JW, Qi SF. et al. Synthetic lethality of combined ULK1 defection and p53 restoration induce pyroptosis by directly upregulating GSDME transcription and cleavage activation through ROS/NLRP3 signaling. J Exp Clin Cancer Res. 2024;43:248
162. Li J, Yang D, Lin Y, Xu W, Zhao SM, Wang C. OTUD3 suppresses the mTORC1 signaling by deubiquitinating KPTN. Front Pharmacol. 2023;14:1337732
163. Nie H, Lin P, Zhang Y, Wan Y, Li J, Yin C. et al. Single-cell meta-analysis of inflammatory bowel disease with scIBD. Nat Comput Sci. 2023;3:522-31
164. Chen J, Xu F, Ruan X, Sun J, Zhang Y, Zhang H. et al. Therapeutic targets for inflammatory bowel disease: proteome-wide Mendelian randomization and colocalization analyses. EBioMedicine. 2023;89:104494
165. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T. et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010;42:1118-25
166. Xu YM, Gao Q, Zhang JZ, Lu YT, Xing DM, Qin YQ. et al. Prolyl hydroxylase 3 controls the intestine goblet cell generation through stabilizing ATOH1. Cell Death Differ. 2020;27:2131-42
167. Lee M, Kim YS, Lim S, Shin SH, Kim I, Kim J. et al. Protein stabilization of ITF2 by NF-kappaB prevents colitis-associated cancer development. Nat Commun. 2023;14:2363
168. Li H, Liang Y, Lai X, Wang W, Zhang J, Chen S. Genetic Deletion of Fbw7 in the mouse intestinal epithelium aggravated dextran sodium sulfate-induced colitis by modulating the inflammatory response of NF-kappaB pathway. Biochem Biophys Res Commun. 2018;498:869-76
169. He J, Song Y, Li G, Xiao P, Liu Y, Xue Y. et al. Fbxw7 increases CCL2/7 in CX3CR1hi macrophages to promote intestinal inflammation. J Clin Invest. 2019;129:3877-93
170. Wang K, Liu F, Muchu B, Deng J, Peng J, Xu Y. et al. E3 ubiquitin ligase RNF180 mediates the ALKBH5/SMARCA5 axis to promote colon inflammation and Th17/Treg imbalance in ulcerative colitis mice. Arch Pharm Res. 2024;47:645-58
171. Rivas MA, Graham D, Sulem P, Stevens C, Desch AN, Goyette P. et al. A protein-truncating R179X variant in RNF186 confers protection against ulcerative colitis. Nat Commun. 2016;7:12342
172. Shen J, Qiao YQ, Ran ZH, Wang TR. Up-regulation and pre-activation of TRAF3 and TRAF5 in inflammatory bowel disease. Int J Med Sci. 2013;10:156-63
173. Ma C, Lin W, Liu Z, Tang W, Gautam R, Li H. et al. NDR1 protein kinase promotes IL-17- and TNF-alpha-mediated inflammation by competitively binding TRAF3. EMBO Rep. 2017;18:586-602
174. Vlantis K, Polykratis A, Welz PS, van Loo G, Pasparakis M, Wullaert A. TLR-independent anti-inflammatory function of intestinal epithelial TRAF6 signalling prevents DSS-induced colitis in mice. Gut. 2016;65:935-43
175. Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M. et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477:330-4
176. Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47:979-86
177. Cleynen I, Juni P, Bekkering GE, Nuesch E, Mendes CT, Schmied S. et al. Genetic evidence supporting the association of protease and protease inhibitor genes with inflammatory bowel disease: a systematic review. PLoS One. 2011;6:e24106
178. Azuara D, Ausso S, Rodriguez-Moranta F, Guardiola J, Sanjuan X, Lobaton T. et al. New Methylation Biomarker Panel for Early Diagnosis of Dysplasia or Cancer in High-Risk Inflammatory Bowel Disease Patients. Inflammatory bowel diseases. 2018;24:2555-64
Author contact
![]() Corresponding authors: Yi Wang, PhD, Professor, E-mail: yi.wang1122edu.cn; Chenjian Zhou, Email: zhouchenjianedu.cn
Corresponding authors: Yi Wang, PhD, Professor, E-mail: yi.wang1122edu.cn; Chenjian Zhou, Email: zhouchenjianedu.cn