Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(9):4618-4632. doi:10.7150/ijbs.124757 This issue Cite
Research Paper
Canagliflozin and Brusatol Synergize against LKB1-KEAP1 Co-Mutant NSCLC through AKT Suppression
Yiguan Chen1*, Xiang-Zheng Gao1*, Dade Rong1, Liangliang Gao1, Mingzhu Tang1, Guang Lu2, Zhi-Qiang Ling3, Han-Ming Shen1 ![]()
1. Faculty of Health Sciences, Ministry of Education Frontier Science Centre for Precision Oncology, University of Macau, Macau SAR, China.
2. Department of Physiological Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, Guangdong, China.
3. Zhejiang Cancer Hospital, Hangzhou, Zhejiang 310022, China; Hangzhou Institute of Medicine (HIM), Chinese Academy of Sciences, Hangzhou, Zhejiang, China.
* These authors contributed equally.
Received 2025-9-5; Accepted 2025-12-18; Published 2026-4-23
Abstract

Liver kinase B1 (LKB1, encoded by STK11) is an important tumour suppressor, with approximately 30% of non-small cell lung cancer (NSCLC) patients harbouring LKB1 mutations. Our previous work showed that LKB1-mutant NSCLC cells are sensitive to glucose starvation, suggesting that suppression of glucose metabolism may serve as a potential therapeutic strategy for NSCLC patients with LKB1 mutation. In this study, we found LKB1 mutations frequently co-occur with mutations in Kelch-like ECH-associated-protein 1 (KEAP1), another key tumour suppressor regulating the NRF2-mediated antioxidant response. To target LKB1-KEAP1 co-mutant NSCLC, we utilized Canagliflozin, an FDA-approved sodium-glucose co-transporter 2 (SGLT2) inhibitor that mimics glucose starvation via inhibiting glucose uptake, in combination with Brusatol, an inhibitor of NRF2 signalling. Our results demonstrate that the combined treatment of Canagliflozin and Brusatol exerts potent anti-tumour effects in LKB1-KEAP1 co-mutant NSCLC cells both in vitro and in vivo. Mechanistically, the combination suppresses AKT activity and promotes AKT degradation, ultimately leading to apoptotic cell death. Taken together, these findings support the potential of combined Canagliflozin and Brusatol treatment as an effective therapeutic approach for LKB1-KEAP1 co-mutant NSCLCs.
Keywords: NSCLC, LKB1, KEAP1, NRF2, Canagliflozin, Brusatol
Introduction
Lung cancer remains one of the most prevalent cancer types globally and is characterized by a high mortality rate. Approximately 85% of lung cancers are histopathologically classified as non-small cell lung cancer (NSCLC) [1]. Based on histological features, the two predominant subtypes of NSCLC are adenocarcinoma (LUAD), which accounts for 40%-50% of cases, and squamous cell carcinoma (LUSC), comprising 20%-30% of cases [2]. Despite significant advancements in therapeutic strategies that have improved clinical outcomes, the five-year survival rate for NSCLC patients remains alarmingly low at less than 20% [2, 3].
Liver kinase B1 (LKB1, encoded by STK11) is a crucial tumor suppressor that regulates cellular metabolism through activation of its direct downstream effector, AMP-activated protein kinase (AMPK), a central energy sensor [4, 5]. LKB1 directly phosphorylates and activates AMPK under metabolic stress. AMPK activation suppresses anabolic metabolism and promotes catabolic processes, including glycolysis and autophagy, to maintain ATP production [6, 7]. In NSCLC, LKB1 is the second most frequently altered tumor suppressor, with loss-of-function mutations occurring in 15-30% of cases across both LUAD and LUSC subtypes [8-11]. These mutations contribute to tumor progression by disrupting energy homeostasis, redox balance, and anti-tumor immunity [6]. Our prior work demonstrated that LKB1-mutant NSCLC cells are highly susceptible to cell death under glucose deprivation. Mechanistically, glucose starvation induces oxidative stress, which causes AMPK oxidation and inactivation in the absence of functional LKB1, ultimately leading to cell death [7].
Canagliflozin is an FDA-approved antihyperglycemic agent for type 2 diabetes that inhibits sodium-glucose co-transporter-2 (SGLT2), thereby blocking renal glucose reabsorption and promoting urinary glucose excretion [12-14]. While SGLT2 is predominantly expressed in renal proximal tubules, accumulating studies have reported elevated SGLT2 levels in metastatic lung cancer cells [15]. Although studies have highlighted the potential anti-tumour effects of SGLT2 inhibitors [16-18], it remains unclear whether SGLT2 inhibitors such as Canagliflozin display selectivity against cancers with specific genetic profiles. Based on our earlier study that LKB1-mutant NSCLCs are highly sensitive to glucose starvation [7, 19], it was important to determine whether Canagliflozin could selectively kill NSCLC cells harboring LKB1 mutation.
Kelch-like ECH-associated-protein 1 (KEAP1) functions as a central regulator of the antioxidant response pathway by acting as a negative regulator of NRF2, a transcription factor essential for cellular adaptation to oxidative stress [20, 21]. Under homeostatic conditions, NRF2 is ubiquitously expressed but maintained at low level through KEAP1-mediated proteasomal degradation [22, 23]. Loss-of-function mutations in KEAP1 occur in approximately 17% of LUAD cases [24]. Disruption of KEAP1-Cullin3 ligase activity leads to NRF2 stabilization and nuclear retention, thereby activating the transcription of antioxidant genes. This adaptive response confers resistance to oxidative damage and apoptosis, highlighting the KEAP1-NRF2 axis as a critical survival mechanism in cancer cells [25].
Brusatol, a natural compound isolated from the medicinal plant Brucea javanica, has been reported to suppress NRF2 activity by promoting its degradation and disrupting antioxidant and detoxification pathways [26-28]. In addition to its effects on NRF2 signalling, Brusatol has shown broader anti-tumour activities in multiple cancer types. For example, in pancreatic cancer, Brusatol has been reported to enhance gemcitabine efficacy by abrogating gemcitabine-induced NRF2 activation and increasing ROS accumulation [28]. In renal cell carcinoma, Brusatol suppresses tumour growth through regulation of the PTEN-PI3K-AKT pathway [29]. In NSCLC, Brusatol induces ROS-mediated mitochondrial apoptosis and enhances radiosensitivity by increasing ROS production and DNA damage [27]. In this study, we systematically evaluated the synergistic anti-tumor efficacy of combined Canagliflozin and Brusatol treatment in LKB1-KEAP1 co-mutant NSCLC models both in vitro and in vivo. Our findings provide preclinical evidence supporting the development of combined Canagliflozin and Brusatol treatment as a potential therapeutic strategy for NSCLC patients with LKB1-KEAP1 co-mutations.
Materials and Methods
Cells and cell culture
A549, H460, H1299, H358, and HEK-293T cells were purchased from American Type Culture Collection (ATCC, USA). Except HEK-293T cells, all cell lines were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco, 11875093) supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco, 10099141C) and 1% (v/v) penicillin/streptomycin (Gibco, 15140122). HEK-293T cells were cultured in Dulbecco's Modified Eagle Medium (DMEM; Gibco, 11965092) supplemented with 10% (v/v) FBS (Gibco, 10099141C) and 1% (v/v) penicillin/streptomycin (Gibco, 15140122). All cell line were maintained at 37°C in a humidified incubator with 5% CO2. For drug-treatment experiments performed under low-glucose conditions, cells were cultured in RPMI 1640 medium containing 5 mM glucose to mimic physiological blood glucose level. 5 mM glucose RPMI 1640 medium was prepared by dissolving D-(+)-glucose (Sigma, G7021) in glucose-free RPMI 1640 medium (Gibco, 11879020).
Reagents and antibodies
The following chemicals were purchased from MedChemExpress: Brusatol (HY-19543), Ferrostatin-1 (HY-100579), GSK-872 (HY-101872A), and hydroxychloroquine (HY-B1370). MG132 (S2619) and Bafilomycin A1 (S1413) were purchased from Selleckchem. Z-VAD (V116) and N-Acetyl-L-cysteine (A9165) were purchased from Sigma-Aldrich. Canagliflozin (A11100) was obtained from Adooq Bioscience. The following primary antibodies were used: AKT (9272, 1:1000), caspase-7 (12827, 1:1000), KEAP1 (8047, 1:1000), LKB1 (3050, 1:1000), PARP (9542, 1:1000), phospho-AKT S473 (4060, 1:1000), and phospho-AMPK T172 (2535, 1:1000) from Cell Signalling Technology; AMPK (10929-2-AP, 1:1000) and 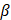 -actin (66009-1-Ig, 1:5000) were purchased from Proteintech; GAPDH (ab8245, 1:5000) and NRF2 (ab137550, 1:1000) from Abcam; ubiquitin (sc-8017, 1:1000) from Santa Cruz Biotechnology.
-actin (66009-1-Ig, 1:5000) were purchased from Proteintech; GAPDH (ab8245, 1:5000) and NRF2 (ab137550, 1:1000) from Abcam; ubiquitin (sc-8017, 1:1000) from Santa Cruz Biotechnology.
Cell viability assay
Cell viability was detected using the propidium iodide exclusion assay (Sigma, P4170). Cells were plated in a 12-well plate one day before treatment. After the designed treatments, medium and cells were all collected by trypsin digestion and centrifuged at 300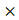 g for 4 minutes at 4°C. The supernatant was discarded, and the pellet was washed with cold PBS twice, then resuspended in the cold PBS containing 5
g for 4 minutes at 4°C. The supernatant was discarded, and the pellet was washed with cold PBS twice, then resuspended in the cold PBS containing 5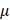 g/ml PI staining reagent and immediately subjected to flow cytometry. Ten thousand cells were analyzed for PI fluorescence intensity using CytoExpert (Beckman Coulter). Data were analyzed using FlowJo_V10 software.
g/ml PI staining reagent and immediately subjected to flow cytometry. Ten thousand cells were analyzed for PI fluorescence intensity using CytoExpert (Beckman Coulter). Data were analyzed using FlowJo_V10 software.
Immunoblotting
Cells were lysed in SDS lysis buffer containing 20% (v/v) glycerol, 2% (w/v) SDS, 62.5mM Tris-HCl (pH6.8), 2mM DTT, supplemented with 1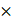 phosphatase and protease inhibitor cocktails. Cell lysates were boiled for 5 minutes, and protein concentration was determined by bicinchoninic acid (BCA) protein assay (Bio-Rad, 5000201). Lysates were mixed with 4
phosphatase and protease inhibitor cocktails. Cell lysates were boiled for 5 minutes, and protein concentration was determined by bicinchoninic acid (BCA) protein assay (Bio-Rad, 5000201). Lysates were mixed with 4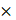 Laemmli loading buffer (Bio-Rad, 1610747). Protein samples (10
Laemmli loading buffer (Bio-Rad, 1610747). Protein samples (10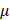 g) were loaded onto SDS-polyacrylamide gels and separated by SDS-PAGE at a constant voltage of 80V, followed by transfer to methanol-activated poly(vinylidene fluoride) (PVDF) membranes at a constant current of 350 mA for 2 hours. Membranes were blocked with SuperBlock T20 (TBS) blocking buffer (Thermo Fisher, ZC386749) for 1 hour at room temperature and then incubated with primary antibodies overnight at 4°C. After primary antibody incubation, membranes were washed three times with Tris-buffered saline containing 0.1% (v/v) TWEEN20 (TBST), with 10 minutes each, and then incubated with the secondary antibodies for 1 hour at room temperature. Chemiluminescence signals were detected using a Bio-Rad ChemiDoc Touch Imaging system. Band intensities were analyzed using Image Lab software.
g) were loaded onto SDS-polyacrylamide gels and separated by SDS-PAGE at a constant voltage of 80V, followed by transfer to methanol-activated poly(vinylidene fluoride) (PVDF) membranes at a constant current of 350 mA for 2 hours. Membranes were blocked with SuperBlock T20 (TBS) blocking buffer (Thermo Fisher, ZC386749) for 1 hour at room temperature and then incubated with primary antibodies overnight at 4°C. After primary antibody incubation, membranes were washed three times with Tris-buffered saline containing 0.1% (v/v) TWEEN20 (TBST), with 10 minutes each, and then incubated with the secondary antibodies for 1 hour at room temperature. Chemiluminescence signals were detected using a Bio-Rad ChemiDoc Touch Imaging system. Band intensities were analyzed using Image Lab software.
ROS measurements
Intracellular ROS levels were measured using CellROX Oxidative Stress Reagent (Thermo Fisher, C10444). Cells were seeded in a 12-well plate. 30 minutes before the end of treatment, CellROX reagent was added to the cells at a final concentration of 5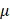 M, followed by incubation at 37°C for 30 minutes. Cells were then washed twice with PBS and harvested by trypsin digestion. Collected cells were centrifugated at 300
M, followed by incubation at 37°C for 30 minutes. Cells were then washed twice with PBS and harvested by trypsin digestion. Collected cells were centrifugated at 300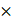 g for 4 minutes at 4°C, and the cell pellet was washed twice with cold PBS. Cells were then resuspended in cold PBS and subjected to flow cytometry (CytoExpert, Beckman Coulter). Data were analyzed using FlowJo_V10 software.
g for 4 minutes at 4°C, and the cell pellet was washed twice with cold PBS. Cells were then resuspended in cold PBS and subjected to flow cytometry (CytoExpert, Beckman Coulter). Data were analyzed using FlowJo_V10 software.
RNA extraction and real time-PCR (RT-PCR)
Cells were seeded in 12-well plate one day before treatment. After treatment, cells were harvested by trypsin digestion followed by centrifugation at 300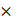 g for 4 minutes. Cell pellet was washed once with PBS and centrifuged again. The pellet was directly subjected to RNA extraction. Total RNA was isolated using the RNAeasy™ Animal RNA Isolation Kit with Spin Column (Beyotime, R0027) according to the manufacturer's instructions. RNA concentration and purity were evaluated by a Nanodrop spectrophotometer. cDNA (1
g for 4 minutes. Cell pellet was washed once with PBS and centrifuged again. The pellet was directly subjected to RNA extraction. Total RNA was isolated using the RNAeasy™ Animal RNA Isolation Kit with Spin Column (Beyotime, R0027) according to the manufacturer's instructions. RNA concentration and purity were evaluated by a Nanodrop spectrophotometer. cDNA (1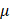 g) was synthesized from the total extracted RNA using the iScript™ cDNA Synthesis Kits (Bio-Rad, 1708891). Real-time PCR was performed using a 10
g) was synthesized from the total extracted RNA using the iScript™ cDNA Synthesis Kits (Bio-Rad, 1708891). Real-time PCR was performed using a 10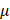 l reaction mixture containing 4
l reaction mixture containing 4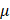 l SsoAdvanced Universal SYBR® Green Supermix (Bio-Rad, 1725274), 1
l SsoAdvanced Universal SYBR® Green Supermix (Bio-Rad, 1725274), 1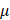 l mixed forward and reverse primers and 200ng cDNA on the Bio-Rad CFX96 Touch Real-Time PCR Detection System. Relative mRNA expression levels were calculated using the 2-
l mixed forward and reverse primers and 200ng cDNA on the Bio-Rad CFX96 Touch Real-Time PCR Detection System. Relative mRNA expression levels were calculated using the 2-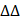 Ct quantification method and normalized to GAPDH. Primer sequences for RT-PCR were as follows:
Ct quantification method and normalized to GAPDH. Primer sequences for RT-PCR were as follows:
Human AKT (Forward: 5'-AGCGACGTGGCTATTGTGAAG-3'; Reverse: 5'-GCCATCATTCTTGAGGAGGAAGT-3');
Human GAPDH (Forward: 5'-CCACTCCTCCACCTTTGACG-3'; Reverse: 5'-CCACCACCCTGTTGCTGTAC-3').
Plasmid construction
To produce retrovirus, pBABE empty vector (Addgene, #1764), pBABE-FLAG-LKB1-WT (Addgene, #8592), and pBABE-FLAG-LKB1-KD (Addgene, #8593) were transfected into Phoenix-AMPHO packaging cells using LipofectamineTM 3000 Transfection Reagent (Thermo Fisher, L3000015) for 48 hours. Retroviral supernatants were collected, centrifugated at 2000  g for 5 minutes, and the supernatant was used to infect A549 and H460 cells in the presence of 5
g for 5 minutes, and the supernatant was used to infect A549 and H460 cells in the presence of 5 g/ml polybrene for 6 hours.
g/ml polybrene for 6 hours.
To produce lentivirus, the KEAP1 construct for overexpression was cloned into the pCDH-CMV vector (Addgene, #72265), and shRNA targeting KEAP1 (Miaoling Biology, P48967) was used for KEAP1 knockdown. HEK-293T cells were seeded in 6-cm dishes at approximately 70% confluency one day before transfection in the DMEM supplemented with 10% (v/v) FBS. Three hours before transfection, the culture medium was replaced with FBS-free DMEM to improve transfection efficacy. PEI MAX-Transfection Grade Linear Polyethyleneimine Hydrochloride (1mg/ml) and lentiviral expression vector were used at a ratio of 3:1, together with 1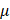 g pCMV-VSV-G and 1
g pCMV-VSV-G and 1 g psPAX2. After 2 hours of transfection, the medium was replaced with DMEM containing 10% (v/v) FBS, and the HEK-293T cells were incubated for an additional 48 hours. Lentiviral supernatants were collected, centrifuged at 2000
g psPAX2. After 2 hours of transfection, the medium was replaced with DMEM containing 10% (v/v) FBS, and the HEK-293T cells were incubated for an additional 48 hours. Lentiviral supernatants were collected, centrifuged at 2000 g for 5 minutes, and the supernatant was added to the pre-seeded target cells for further 48 hours of infection. 2
g for 5 minutes, and the supernatant was added to the pre-seeded target cells for further 48 hours of infection. 2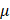 g/ml puromycin (Sigma, P9620) was used to select virally infected cells. Human KEAP1 primer sequences for overexpression were as follows:
g/ml puromycin (Sigma, P9620) was used to select virally infected cells. Human KEAP1 primer sequences for overexpression were as follows:
Forward: 5'-CTGGAGGATCATACCAAGCAGG-3'.
Reverse: 5'-GGATACCCTCAATGGACACCAC-3'.
CRISPR-Cas9-mediated gene knockout
Gene knockouts were performed using single-guide RNAs (sgRNAs) and CRISPR-Cas9 technology. sgRNA oligonucleotides targeting human LKB1 were cloned into the pSpCas9(BB)-2A-Puro (PX459) V2.0 vector. The established sgLKB1 plasmid was transfected into H1299 cells using Lipofectamine 3000 reagent. pLenti-Puro-sgAMPK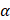 1 (P21236) and pLenti-Puro-sgAMPK
1 (P21236) and pLenti-Puro-sgAMPK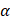 2 (P21235) plasmid were obtained from Miaoling Biology and used to produce lentivirus as described above. Lentiviral particles were added to pre-seeded target cells for 48 hours of infection, and infected cells were selected with 2
2 (P21235) plasmid were obtained from Miaoling Biology and used to produce lentivirus as described above. Lentiviral particles were added to pre-seeded target cells for 48 hours of infection, and infected cells were selected with 2 g/ml puromycin for 1 week. Monoclonal cells were then selected and maintained in medium containing 1
g/ml puromycin for 1 week. Monoclonal cells were then selected and maintained in medium containing 1 g/ml puromycin. Positive monoclonal cells with LKB1 knockout or AMPK
g/ml puromycin. Positive monoclonal cells with LKB1 knockout or AMPK  1/
1/  2 double knockout were verified by immunoblotting. The human LKB1 sgRNA sequences were as follows:
2 double knockout were verified by immunoblotting. The human LKB1 sgRNA sequences were as follows:
Forward: 5'-CACCGTTGCGAAGGACTCCCCAACG-3'.
Reverse: 5'-AAACCGTTGGGGATCCTTCGCAAC-3'.
Tumour xenograft studies
Male BALB/c nude mice (4-5 weeks old) were obtained from the University of Macau Animal Facility. Mice were randomly divided into four groups and treated with vehicle, Invokana (clinical formulation of Canagliflozin; 200 mg/kg/day), Bursatol (2 mg/kg/day), or the combination of both drugs. Invokana was administered via oral gavage, and Brusatol was delivered intraperitoneally every three days for two weeks. A549 or H1299 (5 × 106 cells in 180 l PBS) cells were subcutaneously implanted into the dorsal flanks of the nude mice. Tumour volume and mouse weight were recorded by callipers and a weighing scale, respectively. Tumour volume was calculated using the formula (a² × b × 0.5), where a and b represent the shortest diameter and longest perpendicular diameters, respectively. At the end of treatments, mice were euthanized by CO₂ asphyxiation. Excised xenograft tissues were weighed, frozen in liquid nitrogen, and stored at -80°C for subsequent analysis.
l PBS) cells were subcutaneously implanted into the dorsal flanks of the nude mice. Tumour volume and mouse weight were recorded by callipers and a weighing scale, respectively. Tumour volume was calculated using the formula (a² × b × 0.5), where a and b represent the shortest diameter and longest perpendicular diameters, respectively. At the end of treatments, mice were euthanized by CO₂ asphyxiation. Excised xenograft tissues were weighed, frozen in liquid nitrogen, and stored at -80°C for subsequent analysis.
Statistical analysis
All experimental procedures were independently repeated at least three times with consistent results. Statistical analyses were conducted using GraphPad Prism software (v7.0), and the corresponding p-values are reported in the figure panels and figure legends. A p-value of < 0.05 was considered statistically significant.
Results
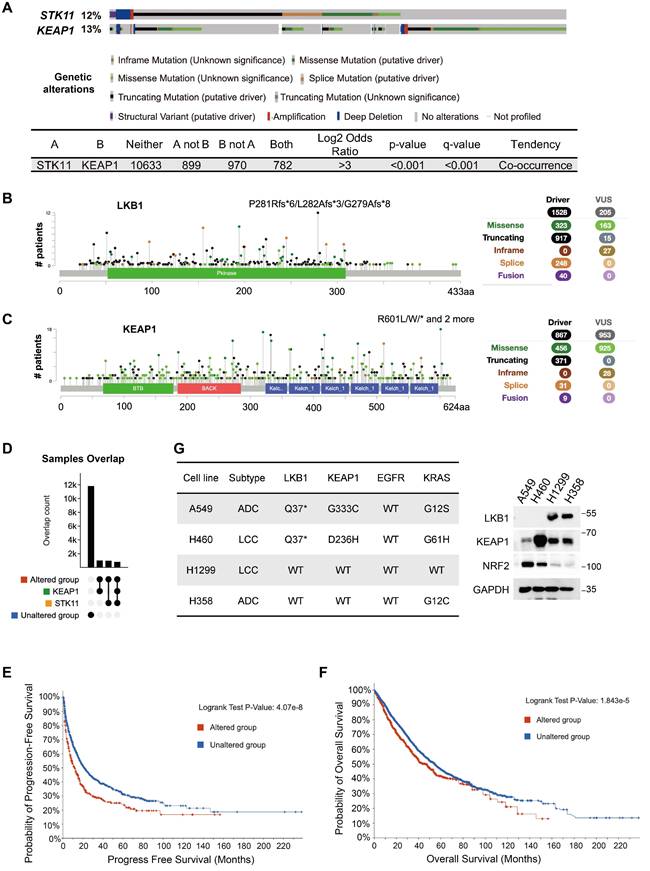
Co-occurrence of STK11 and KEAP1 mutations in NSCLC
To investigate the genetic relationship between STK11 and KEAP1 mutations, we analyzed genomic data from 30 studies in cBioPortal, comprising 14,700 samples from 12,261 patients. Somatic mutations in STK11 (12%) and KEAP1 (13%) were observed at comparable frequencies across the cohorts (Figure 1A). The predominant mutations in STK11 were truncating (60%), followed by missense (21.2%) and splice site (16.2%) variants (Figure 1B). In contrast, KEAP1 alterations consisted mainly of missense (52.6%) and truncating (42.5%) mutations (Figure 1C). Strikingly, among the 2,876 samples harbouring at least one alteration in STK11 or KEAP1 (termed the altered group), approximately 30% (884 samples) exhibited co-mutations in both genes (Figure 1A and 1D). The prevalence of samples with dual mutations was comparable to that of samples with single-gene mutations, suggesting a pronounced tendency for STK11 and KEAP1 co-mutation. Next, we performed survival analyses in patients with STK11-KEAP1 co-mutations. In the altered group, both progression-free survival (PFS) (p = 4.07e-8 < 0.0001) and overall survival (OS) (p = 1.843e-5 < 0.0001) were significantly reduced compared to the non-altered group (Figure 1E and 1F). Specifically, the median PFS was 11.64 months (95% CI: 9.01-14.66) in the altered group and 21.07 months (95% CI: 18.61-24.99) in the non-altered group, while the median OS was 45.57 months (95% CI: 39.3-51.27) and 54.4 months (95% CI: 50.89-56.71), respectively (Figure 1E and 1F). These results indicated that STK11-KEAP1 co-mutations were associated with a more aggressive disease phenotype, with poorer prognosis. To validate these findings in cancer cell lines, we examined the basal expression levels of LKB1, KEAP1, and NRF2 in four NSCLC cell lines. H1299 and H358 cells retained wildtype LKB1 and KEAP1, whereas A549 and H460 cells were LKB1-deficient, consistent with truncating STK11 mutations. In addition, although KEAP1 protein expression was preserved in A549 and H460 cells, the G333C and D236H mutations impaired the ability of KEAP1 to bind NRF2 and promote its proteasomal degradation, resulting in constitutive NRF2 activation (Figure 1G).
Co-occurrence of STK11 and KEAP1 mutations in NSCLC. 30 reported NSCLC studies (16 on LUAD, 6 on LUSC, and 8 on both) with a total of 14,700 samples from 12261 patients were pooled for analysis. (A) The mutation spectrum of STK11 and KEAP1. (B and C) Mutation type, frequency, and localization within the protein structure of (B) LKB1 and (C) KEAP1. Circles in distinct colours represent different mutation types. (D) Overlap counts of STK11 and KEAP1 mutations in the pooled samples. (E and F) Kaplan-Meier curves showing (E) progression-free survival and (F) overall survival in the entire cohort, comparing samples without mutations in STK11 or KEAP1 (unaltered group) with samples harbouring at least one mutation in either gene (altered group). (G) Expression and mutation status of the indicated proteins in four NSCLC cell lines (ADC: adenocarcinoma; LCC: large cell carcinoma).
Canagliflozin and Brusatol selectively target LKB1-KEAP1 co-mutant NSCLC
To evaluate the potential cytotoxic effects of Canagliflozin and Brusatol, we examined cell survival in a panel of NSCLC cell lines treated with increasing concentrations of each compound. Single treatment with either Canagliflozin or Brusatol induced modest cytotoxicity in LKB1-KEAP1 wildtype cells (H1299 and H358), with cell viability remaining above 80% after 48 hours. In contrast, LKB1-KEAP1 co-mutant cell lines (A549 and H460) exhibited concentration-dependent reductions in survival, with H460 cells showing greater sensitivity than A549 cells at equivalent doses (Figure 2A and 2B). These results indicated that treatment with either Canagliflozin or Brusatol alone selectively killed LKB1-KEAP1 co-mutant NSCLC cells in a dose-dependent manner.
Canagliflozin and Brusatol selectively target LKB1-KEAP1 co-mutant NSCLC. (A and B) Cell viability was examined by PI exclusion assay following treatment with (A) Canagliflozin or (B) Brusatol for 48 hours in four NSCLC cell lines. (C and D) Immunoblot results of AMPK activity under Canagliflozin treatment (0, 10, 25, 50, 75, 100 μM) for 12 hours in (C) LKB1-KEAP1 wildtype and (D) LKB1-KEAP1 co-mutant cells. (E) Immunoblot results of AMPK activity under Canagliflozin treatment for 12 hours with different status of LKB1. (F) Immunoblot analysis of NRF2 activity under Brusatol treatment (0.1, 0.25, 0.5, 1, 5 μM) for 6 hours in LKB1-KEAP1 co-mutant cells. (G) Effect of MG132 on Brusatol-treated A549 cells. Cells were pretreated with MG132 (10 μM) for 2 hours, followed by treatment with 500 nM Brusatol treatment for the indicated timepoints. **p < 0.01, ***p < 0.001, ****p < 0.0001 by unpaired t-test. ★ indicates non-specific bands.
To determine the mechanism underlying the effects of Canagliflozin and Brusatol, we first examined the changes in the LKB1-AMPK pathway. In LKB1 wildtype cells (H1299 and H358), Canagliflozin robustly enhanced AMPK activity (Figure 2C), consistent with its ability to mimic glucose deprivation by blocking glucose uptake. Conversely, LKB1-mutant cells (A549 and H460) displayed reduced AMPK phosphorylation following Canagliflozin treatment (Figure 2D). High doses of Canagliflozin further decreased total AMPK protein levels in H460 cells (Figure 2D), similar to our previous report under glucose starvation [7]. Re-expression of wildtype (WT) LKB1, but not empty vector (EV), in A549 cells restored AMPK phosphorylation, whereas LKB1 knockout (KO) in H1299 cells abolished AMPK activation in response to Canagliflozin treatment (Figure 2E). These results thus demonstrated that the high sensitivity of LKB1-mutant cells to Canagliflozin was mediated via AMPK inactivation.
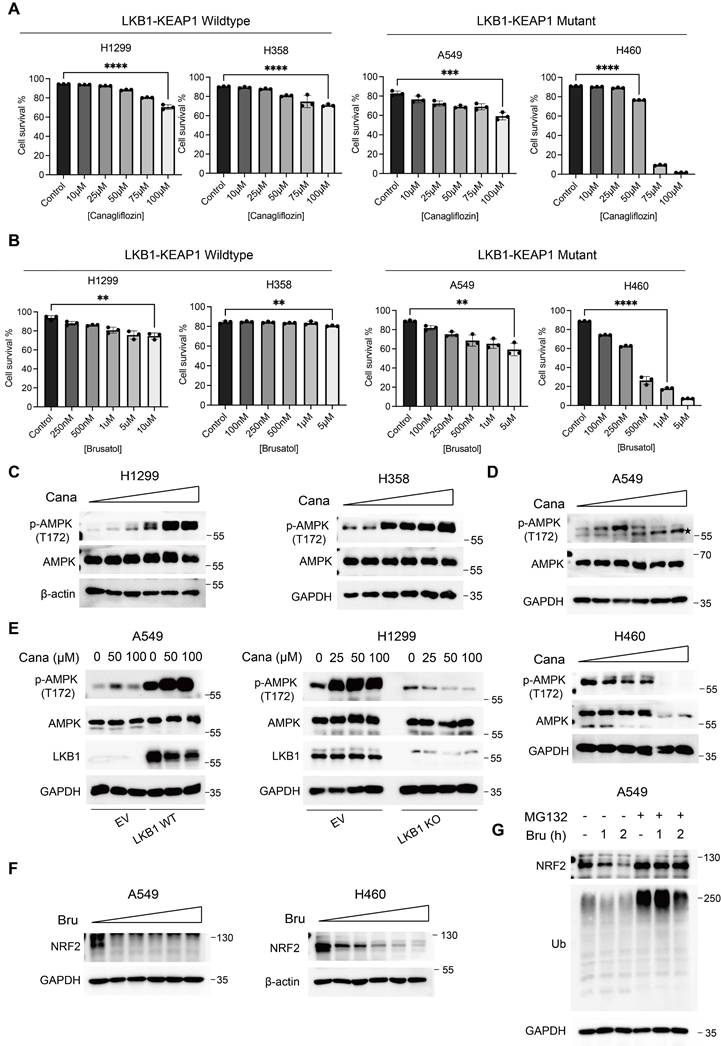
Based on the fact that KEAP1 mutation leads to constitutive NRF2 activation, we next assessed the effects of the NRF2 inhibitor, Brusatol, in KEAP1-mutant NSCLC cells [20]. Brusatol reduced NRF2 protein levels at nanomolar concentrations, however, this effect was transient, with NRF2 expression re-appears after prolonged treatment [26]. We therefore employed 1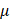 M Brusatol in subsequent experiments (Figure 2F). Pretreating KEAP1-mutant A549 cells with the proteasome inhibitor MG132 reversed the Brusatol-induced NRF2 downregulation (Figure 2G), suggesting that Brusatol promotes NRF2 degradation through ubiquitination and proteasomal degradation mediated by E3 ligases other than KEAP1. The precise mechanism remains to be elucidated in future studies.
M Brusatol in subsequent experiments (Figure 2F). Pretreating KEAP1-mutant A549 cells with the proteasome inhibitor MG132 reversed the Brusatol-induced NRF2 downregulation (Figure 2G), suggesting that Brusatol promotes NRF2 degradation through ubiquitination and proteasomal degradation mediated by E3 ligases other than KEAP1. The precise mechanism remains to be elucidated in future studies.
Synergistic effects of Canagliflozin and Brusatol in LKB1-KEAP1 co-mutant NSCLC
To further determine whether combined treatment with Canagliflozin and Brusatol exerts a synergistic effect, we treated A549, H460, H1299, and H358 cells with either single agents or the combination and assessed cell viability by PI exclusion assay. LKB1-KEAP1 co-mutant cell lines (A549 and H460) exhibited significant cell death following combined treatment with Canagliflozin (75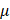 M) and Brusatol (1
M) and Brusatol (1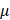 M) (Figure 3A). The synergistic effect of the combination was further confirmed by the Combination Index analysis [30, 31]. In contrast, no obvious synergistic effect was observed in LKB1-KEAP1 wildtype cells (H1299 and H358) under the same treatment conditions (Figure 3B). Given the frequent co-occurrence of LKB1 and KEAP1 mutations in NSCLC, we next examined the effects of Canagliflozin and Brusatol in NSCLC cells carrying single mutations in either LKB1 or KEAP1 using genetic manipulation approaches. Restoration of WT KEAP1 in A549 cells reduced NRF2 levels, whereas KEAP1 knockdown (shKEAP1) in H1299 cells elevated NRF2 expression (Figure 3C). Brusatol alone or in combination with Canagliflozin elicited pronounced cytotoxicity in cells with KEAP1 dysregulation than in their wildtype counterparts. Conversely, Canagliflozin alone caused comparable cytotoxic effects regardless of KEAP1 status (Figure 3D). Given that KEAP1 regulates NRF2 expression via ubiquitination and proteasomal degradation, restoration of KEAP1 could be expected to confer cytoprotection, whereas KEAP1-mutant cells exhibited heightened sensitivity to Brusatol-mediated NRF2 inhibition.
M) (Figure 3A). The synergistic effect of the combination was further confirmed by the Combination Index analysis [30, 31]. In contrast, no obvious synergistic effect was observed in LKB1-KEAP1 wildtype cells (H1299 and H358) under the same treatment conditions (Figure 3B). Given the frequent co-occurrence of LKB1 and KEAP1 mutations in NSCLC, we next examined the effects of Canagliflozin and Brusatol in NSCLC cells carrying single mutations in either LKB1 or KEAP1 using genetic manipulation approaches. Restoration of WT KEAP1 in A549 cells reduced NRF2 levels, whereas KEAP1 knockdown (shKEAP1) in H1299 cells elevated NRF2 expression (Figure 3C). Brusatol alone or in combination with Canagliflozin elicited pronounced cytotoxicity in cells with KEAP1 dysregulation than in their wildtype counterparts. Conversely, Canagliflozin alone caused comparable cytotoxic effects regardless of KEAP1 status (Figure 3D). Given that KEAP1 regulates NRF2 expression via ubiquitination and proteasomal degradation, restoration of KEAP1 could be expected to confer cytoprotection, whereas KEAP1-mutant cells exhibited heightened sensitivity to Brusatol-mediated NRF2 inhibition.
Synergistic effects of Canagliflozin and Brusatol in LKB1-KEAP1 co-mutant NSCLC. (A and B) Cell viability was examined by PI exclusion assay following the combination treatment of Canagliflozin (75 μM) and Brusatol (1 μM) for 48 hours in (A) LKB1-KEAP1 co-mutant cells and (B) LKB1-KEAP1 wildtype cells. (C and E) Immunoblot analysis of cells with different status of (C) KEAP1 and (E) LKB1. (D and F) cell viability was measured in cells with different status of (D) KEAP1 and (F) LKB1 treated with Canagliflozin (75 μM or Brusatol (1 μM) for 48 hours. **p < 0.01, ***p < 0.001, ****p < 0.0001 by unpaired t-test (A and B), or two-way ANOVA test (D and F).
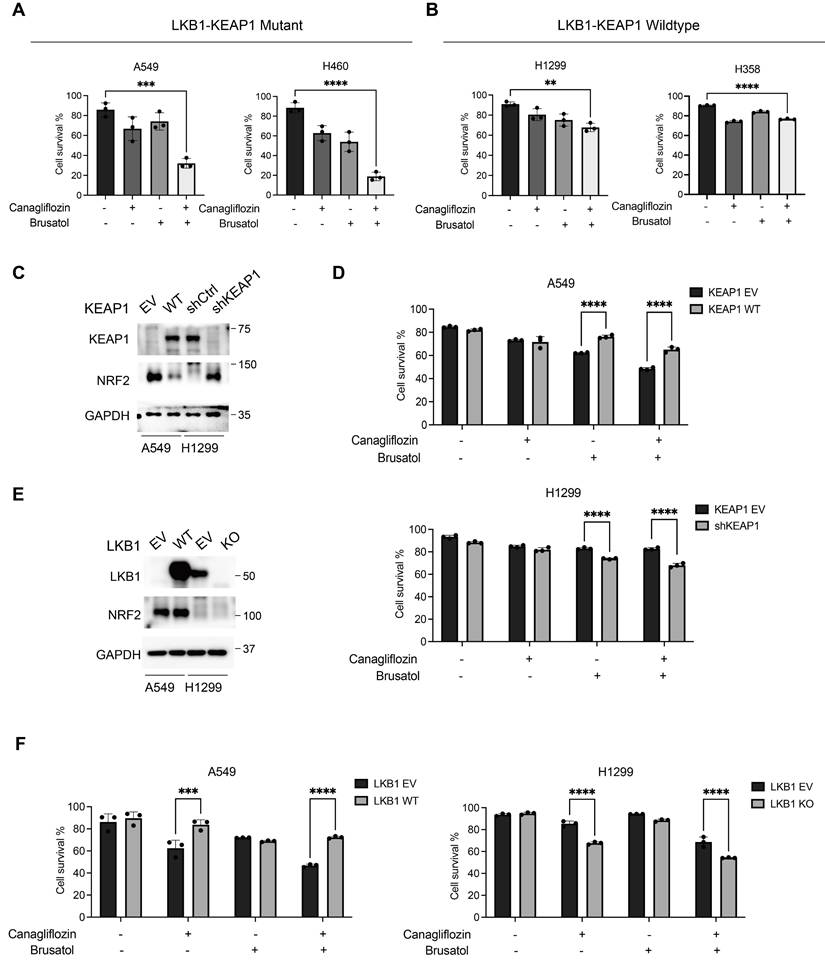
To assess whether a reciprocal relationship exists between LKB1 and NRF2, we used opposite approaches to manipulate LKB1 status by reconstituting WT LKB1 in A549 cells and deleting LKB1 in H1299 cells. Neither intervention altered NRF2 expressions, indicating that LKB1 does not directly regulate NRF2 (Figure 3E). Although LKB1-deficient cells were highly sensitive to Canagliflozin alone or in combination with Brusatol, LKB1 status did not affect sensitivity to Brusatol alone (Figure 3F). These findings demonstrated that the efficacy of Brusatol depends on the KEAP1-NRF2 axis but is independent of LKB1, whereas the activity of Canagliflozin depends on LKB1 status rather than KEAP1.
Combination treatment of Canagliflozin and Brusatol induces apoptosis in LKB1-KEAP1 co-mutant NSCLC
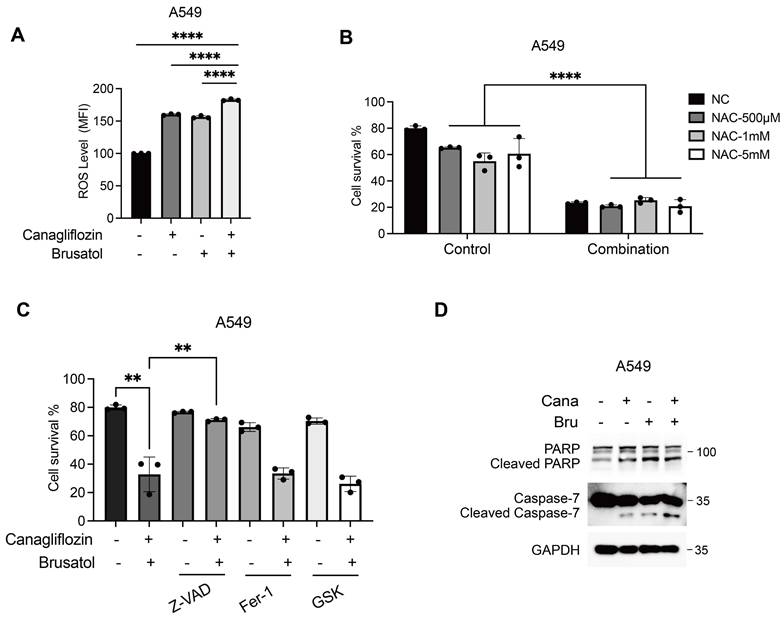
In KEAP1-mutant cells, constitutive activation of NRF2 drives transcriptional upregulation of antioxidant genes, reducing intracellular ROS levels and protecting cancer cells from oxidative stress [32, 33]. Brusatol counteracts this cytoprotective mechanism by promoting NRF2 ubiquitination and proteasomal degradation, thereby exacerbating ROS accumulation [26]. Similarly, glucose starvation has been shown to increase ROS levels, sensitizing LKB1-mutant NSCLC cells through AMPK oxidation and inactivation [7]. To investigate the molecular mechanism underlying the synergistic cytotoxicity of Canagliflozin and Brusatol in LKB1-KEAP1 co-mutant NSCLC cells, we measured intracellular ROS levels using the CellROX assay, which primarily detects superoxide. Treatment with either Canagliflozin or Brusatol alone increased the ROS levels compared with the untreated control, while the combination treatment produced a stronger effect (Figure 4A). To further evaluate the functional role of ROS in the observed cytotoxicity, cells were pretreated with the ROS scavenger N-acetylcysteine (NAC). Unexpectedly, despite testing multiple concentrations, NAC failed to rescue cell death induced by the combination treatment, suggesting the synergistic effect of these two agents operates via another mechanism (Figure 4B). To delineate the type of cell death induced by co-treatment, we employed pharmacological inhibitors targeting distinct cell death pathways, including the pan-caspase inhibitor Z-VAD, the ferroptosis inhibitor ferrostatin-1, and the necroptosis inhibitor GSK-872. Pretreatment with Z-VAD, but not ferrostatin-1 or GSK-872, significantly attenuated cell death induced by the combined treatment, indicating that apoptosis is the predominant mechanism (Figure 4C). This conclusion was further supported by increased levels of cleaved PARP and cleaved caspase 7, two apoptotic hallmarks, in cells treated with both Canagliflozin and Brusatol (Figure 4D). Taken together, these results suggest that the synergistic cytotoxicity of Canagliflozin and Brusatol in LKB1-KEAP1 co-mutant NSCLC cells is mediated predominantly by apoptosis rather than by ROS accumulation alone.
Combination treatment of Canagliflozin and Brusatol induces apoptosis in LKB1-KEAP1 co-mutant NSCLC. (A) Intracellular ROS levels in A549 cells following single or combined treatment with Canagliflozin (75 μM) and Brusatol (1 μM) in A549 for 18 hours. (B) Effect of NAC on combination treatment-induced cell death. Cells were pretreated with the indicated concentration of NAC for 2 hours, followed by combination treatment for 48 hours. (C) Effects of various cell death inhibitors on cytotoxicity induced by the combination treatment. Cells were pretreated with Z-VAD (40 μM), Ferrastatin-1 (10 μM), or GSK-872 (10 μM) for 2 hours, followed by combination treatment for 48 hours, and cell viability was quantified. (D) Immunoblot analysis of apoptotic markers under combination treatment for 24 hours. **p < 0.01 and ****p < 0.0001 by unpaired t-test (A and C) or two-way ANOVA test (B).
Apoptosis induced by Canagliflozin and Brusatol is mediated by suppression of AKT activity in LKB1-KEAP1 co-mutant NSCLC
Both Canagliflozin and Brusatol have been implicated in modulating the PI3K/AKT/mTOR signalling axis to influence tumorigenesis [29, 34]. Brusatol, for instance, suppressed renal cancer cell proliferation by upregulating PTEN, a key tumour suppressor with lipid and protein phosphatase activities, thereby inhibiting the PI3K-AKT pathway [29]. In LKB1-KEAP1 co-mutant cells, treatment with either Canagliflozin or Brusatol alone significantly reduced both phosphorylated AKT and total AKT levels, whereas the combined treatment further suppressed AKT activity (Figure 5A). In contrast, LKB1-KEAP1 wildtype cells showed minimal changes in phosphorylated AKT and total AKT levels following the co-treatment (Figure 5B). Given the established role of AKT as a master regulator of cell survival and anti-apoptotic signalling [35, 36], its inhibition in LKB1-KEAP1 co-mutant cells following treatment with Canagliflozin and Brusatol suggests that the suppression of this anti-apoptotic pathway is a primary driver of their high susceptibility to the combination treatment.
Apoptosis induced by Canagliflozin and Brusatol is mediated by suppression of AKT activity in LKB1-KEAP1 co-mutant NSCLC. (A and B) Immunoblot analysis of AKT activity under the combination treatment for 12 hours in (A) LKB1-KEAP1 co-mutant cells and (B) LKB1-KEAP1 wildtype cells. (C and D) Immunoblot results of AKT activity under Canagliflozin treatment for 12 hours with different status of (C) LKB1 and (D) AMPK. (E and F) Immunoblot results of AKT activity in H1299 cells with different status of (E) LKB1 or (F) KEAP1. (G) Effects of various protein degradation pathway inhibitors on AKT downregulation induced by the combination treatment. Cells were pretreated with MG132 (10 μM), Bafilomycin A1 (BalfA1, 100 μM), or hydroxychloroquine (HCQ, 20 mM) for 2 hours, followed by combination treatment for 12 hours. (H) RT-PCR analysis of AKT mRNA levels in H460 cells upon single-agent or combined treatments for 12 hours. ***p < 0.001 by unpaired t-test.
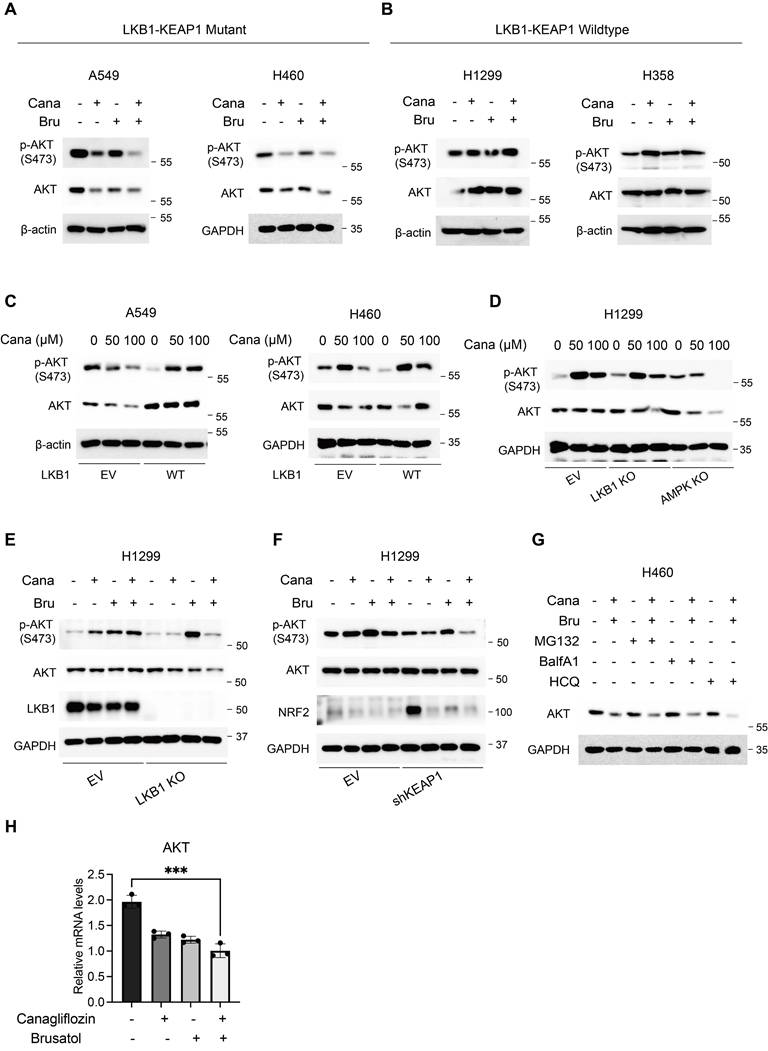
Next, we further elaborated on the respective roles of LKB1 and KEAP1 in regulating AKT activity. Restoration of functional LKB1 in A549 and H460 cells, both of which harbour LKB1-KEAP1 co-mutations, reversed the Canagliflozin-induced suppression of AKT activity, as evidenced by recovery of both phosphorylated and total AKT levels (Figure 5C). The reduction in phosphorylated AKT was consistent with the decline in phosphorylated AMPK observed following Canagliflozin treatment (Figure 2E). Similarly, genetic deletion of LKB1 or its downstream effector AMPK in H1299 cells attenuated AKT activation in response to Canagliflozin (Figure 5D), highlighting the requirement for an intact LKB1-AMPK axis in maintaining AKT activity. Furthermore, combined treatment with Canagliflozin and Brusatol strongly suppresses AKT activity in H1299 cells depleted of either LKB1 or KEAP1 (Figure 5E and 5F). Collectively, these findings demonstrated that both LKB1 and KEAP1 are required to maintain AKT activity.
Finally, we sought to clarify the mechanism of AKT degradation during combination treatment. It is interesting to note that neither proteasome inhibitor MG132 nor the lysosomal inhibitors bafilomycin A1 and hydroxychloroquine rescued the reduction in AKT protein levels induced by the co-treatment of Canagliflozin and Brusatol, indicating that AKT downregulation is independent of the canonical ubiquitin-proteasomal or lysosomal degradation systems (Figure 5G). To test the possibility that AKT reduction might instead occur at the transcriptional level, we measured AKT mRNA levels. As expected, single treatment with either Canagliflozin or Brusatol largely reduced AKT mRNA levels, and combination treatment further exacerbated this effect (Figure 5H). Together, these data indicated that the reduction in total AKT protein observed during combination treatment was driven by transcriptional downregulation of AKT. Future studies are needed to define the precise mechanisms controlling AKT transcription under this setting.
Combination treatment of Canagliflozin and Brusatol exerts anti-tumour effects in the xenograft model in vivo
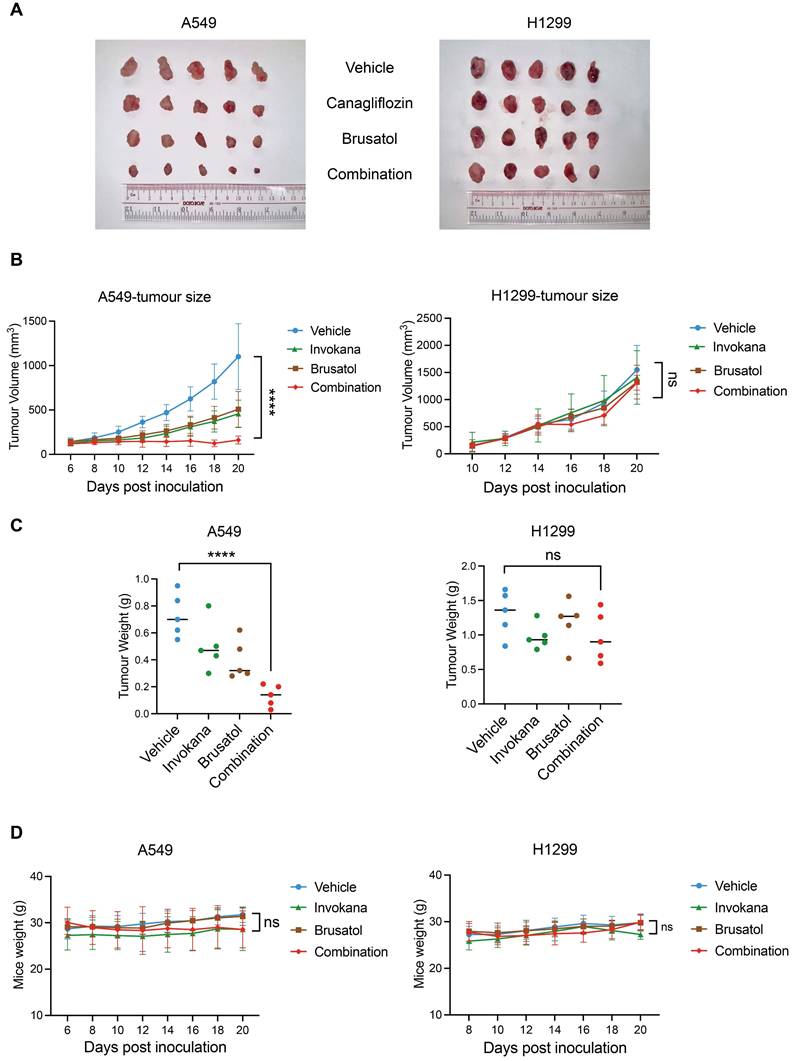
To evaluate the anti-tumour efficacy of Canagliflozin and Brusatol co-treatment in vivo, we established xenograft models using male BALB/c nude mice implanted with A549 (LKB1-KEAP1co-mutant) or H1299 (LKB1-KEAP1 wildtype) cells. Mice were randomly and unbiasedly allocated into four groups: vehicle, Invokana (clinical formulation of Canagliflozin; 200 mg/kg/day), Brusatol (2 mg/kg/day), or the combination treatment. In A549 xenografts, treatment with either Invokana or Brusatol alone modestly suppressed tumour growth, whereas the combination treatment induced tumour regression, as evidenced by a substantial reduction in tumour volume (Figure 6A and 6B). In contrast, neither single-treatment nor the combination treatment reduced tumour growth in H1299 xenografts (Figure 6A and 6B). Final tumour weights were consistent with the tumour volume (Figure 6C). Notably, no significant loss of body weight was observed in any treatment group (Figure 6D), indicating minimal systemic toxicity. These results thus demonstrated that co-treatment with Invokana and Brusatol exerts selective therapeutic efficacy in LKB1-KEAP1 co-mutant NSCLC in vivo.
Combination treatment of Canagliflozin and Brusatol exerts anti-tumour effects in the xenograft model in vivo. (A) Images of A549- and H1299-derived xenograft tumours at the endpoint of treatment with vehicle, single-agent treatment, or combined treatments with Invokana (200 mg/kg/day) and Brusatol (2 mg/kg/day). (B) Tumour volume measurements over the course of the treatment in A549 and H1299 xenografts (A549: n=5; H1299: n=5). (C) Final tumour weights of A549- and H1299-derived xenografts under the treatment conditions shown in (A). (D) Body weight measurements of mice during the treatment period. ****p < 0.0001 by two-way ANOVA test (B and D), or unpaired t-test (C). Quantitative data are represented as mean ± SD (B and D) or medium (C).
Discussion
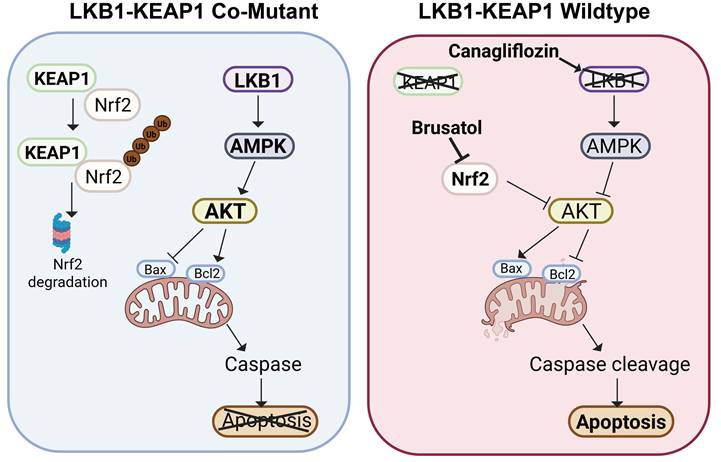
LKB1 (encoded by STK11) and KEAP1 are two vital tumour suppressors in NSCLC. Patients with NSCLC harbouring LKB1 or KEAP1 mutations are often less responsive to current targeted therapies, including EGFR inhibitors [37, 38]. Therefore, the development of new therapeutic strategies for NSCLC patients with these genetic alterations remain an important unmet need. In this study, we used four well-characterized NSCLC cell lines with distinct LKB1 and KEAP1 genotypes to model an aggressive molecular subset of human NSCLC. We first confirmed the co-occurrence of LKB1 and KEAP1 mutations in NSCLC and then evaluated two pharmacological agents, Canagliflozin, a selective SGLT2 inhibitor, and Brusatol, an NRF2 inhibitor, as a therapeutic strategy for LKB1-KEAP1 co-mutant NSCLC. Our data showed that combined treatment with Canagliflozin and Brusatol selectively killed NSCLC cells harbouring LKB1-KEAP1 co-mutations, both in vitro and in vivo, by suppressing AKT activity and ultimately inducing apoptosis (as summarized in Figure 7). Taken together, our findings identify a potential therapeutic strategy for this particularly aggressive molecular subtype and may lay the foundation for future translational clinical studies.
Schematic model of Canagliflozin and Brusatol combination treatment selectively mediate anti-tumour effects in LKB1-KEAP1 co-mutant NSCLC. In normal LKB1-KEAP1 wildtype cells, NRF2 is constitutively degraded by KEAP1 via proteosomal degradation. Under stress conditions, intact AKT signalling prevents apoptosis in the presence of functional LKB1 and AMPK. In LKB1-KEAP1 co-mutant NSCLC cells, Canagliflozin and Brusatol individually suppress AMPK activity and promote NRF2 degradation, respectively. Combined treatment with both drugs synergistically induces cell apoptosis through inhibition of AKT activity.
Canagliflozin, a selective SGLT2 inhibitor, was approved by the FDA in 2013 as an oral antihyperglycemic agent for the treatment of type 2 diabetes mellitus (T2DM). Recently, accumulating evidence has suggested that Canagliflozin exerts biological effects beyond glycaemic control, including cardiovascular and renal protection [12]. Notably, unlike the other SGLT2 inhibitors Dapagliflozin and Empagliflozin, Canagliflozin has been reported to inhibit mitochondrial electron transport chain (ETC) complex I, thereby reducing oxidative phosphorylation and ATP production and potentially increasing mitochondrial ROS levels [39]. Given that high SGLT2 expression has been reported in both lung premalignant cells and metastatic lung cancer lesions, targeting SGLT2 with Canagliflozin may represent a potential therapeutic strategy in NSCLC [19]. In line with this possibility, our data show that Canagliflozin selectively kills LKB1-mutant NSCLC cells. Mechanistically, in the absence of LKB1, Canagliflozin mimics the effect of glucose starvation, resulting in the inactivation of AMPK and AKT, and ultimately leading to apoptotic cell death. These findings support the notion of repurposing Canagliflozin as a cancer therapeutic agent for NSCLC with LKB1 mutations.
As another key tumour suppressor in NSCLC, KEAP1 negatively regulates NRF2 through the Cullin 3-based E3 ubiquitin ligase complex. Mutation of KEAP1 disrupts this regulatory mechanism, leading to stabilization and aberrant activation of NRF2, a major transcriptional factor involved in antioxidant defence and cell survival [40]. It is noteworthy that NSCLC cells harbouring KEAP1 mutations often become highly dependent on NRF2 signalling for tumour maintenance, a phenomenon termed 'NRF2 addiction' [41]. This concept provides the rationale for testing Brusatol as a therapeutic agent in KEAP1-mutant NSCLC. Previous studies have demonstrated that Brusatol exerts pro-apoptotic activity in multiple cancer models, including NSCLC and pancreatic adenocarcinoma [27, 28]. However, the anti-cancer function of Brusatol has not been clearly linked to specific genetic backgrounds. In this study, we found that Brusatol selectively killed NSCLC cells with KEAP1 mutation, further supporting the therapeutic potential of genotype-based treatment in this context.
A hallmark metabolic feature of cancer cells is the Warburg effect, which refers to the preferential reliance on aerobic glycolysis [42-44]. Recent studies have attempted to develop targeted therapeutic strategies for LKB1-KEAP1 co-mutant tumours on the basis of their metabolic vulnerabilities. It has been reported that concurrent loss of LKB1 and KEAP1 promotes metabolic reprogramming from glycolytic dependence towards glutaminolysis [45, 46]. In addition, targeting glutaminase, which converts glutamine to glutamate, has been shown to sensitize LKB1-KEAP1 co-mutant cells to radiotherapy [37]. Thus, our study provides a new line of evidence that combined treatment with Canagliflozin and Brusatol may represent an effective strategy for NSCLC with LKB1-KEAP1 co-mutations.
In summary, our study proposes a novel targeted therapeutic strategy for NSCLC harbouring LKB1-KEAP1 co-mutations through the combined use of Canagliflozin and Brusatol. This new therapeutic approach may represent a potential treatment option for patients with this aggressive molecular subtype. Nevertheless, several limitations of this study should be noted. Although we showed that combined Canagliflozin and Brusatol treatment exerted anti-tumour effects in NSCLC cell lines and xenograft models, the study was based on a limited number of preclinical systems. Further validation in the patient-derived xenografts (PDXs) will be important, as this model better reflects the complex heterogeneity of human tumours. In addition, the upstream mechanism linking LKB1-KEAP1 co-mutation to AKT regulation was not fully elucidated. In particular, although AKT mRNA levels were reduced, the transcriptional regulators responsible for this effect were not investigated in the present study. Therefore, future studies will be needed to clarify this mechanism, which will be important for strengthening the translational potential of this therapeutic approach.
Acknowledgements
Funding
This work was supported by the following research grants to HMS from the University of Macau (CPG2024-0035-FHS, CPG2025-0031-FHS) and the Macau Science and Technology Development Fund (FDCT0031/2021/A1, FDCT0081/2022/AMJ, FDCT0036/2024/RIB1), and the National Key R&D Program of China (No. 2022YFE0205900) to ZQL. YC, XZG, DR, LG and MT were supported by assistantship from the University of Macau. We also thank the Animal Research Core from the University of Macau for the technical support and assistance.
Ethics statement
Animal study was approved by the Ethics Committee of University of Macau (Approved protocol ID: UMARE-009-2022).
Availability data and materials
All data supporting the findings of this study are available from the corresponding author upon reasonable request.
Author contributions
YC and HMS designed the project, performed the experiments, analyzed and interpreted the data, and drafted the manuscript. XZG, DR, LG, and MT performed the experiments and analyzed the data. GL and ZQL analyzed and interpreted the data, and ZQL in resource acquisition. YC and HMS revised the manuscript, and all authors have approved the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71:7-33
2. Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JHM, Beasley MB. et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J Thorac Oncol. 2015;10:1243-60
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30
4. Hardie DG, Alessi DR. LKB1 and AMPK and the cancer-metabolism link - ten years after. BMC Biol. 2013;11:36
5. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563-75
6. Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA. et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329-35
7. Ren Y, Chen J, Chen P, Hao Q, Cheong LK, Tang M. et al. Oxidative stress-mediated AMPK inactivation determines the high susceptibility of LKB1-mutant NSCLC cells to glucose starvation. Free Radic Biol Med. 2021;166:128-39
8. Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K. et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069-75
9. Matsumoto S, Iwakawa R, Takahashi K, Kohno T, Nakanishi Y, Matsuno Y. et al. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene. 2007;26:5911-8
10. Shire NJ, Klein AB, Golozar A, Collins JM, Fraeman KH, Nordstrom BL. et al. STK11 (LKB1) mutations in metastatic NSCLC: Prognostic value in the real world. PLoS One. 2020;15:e0238358
11. Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R. et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893-8
12. Mahaffey KW, Jardine MJ, Bompoint S, Cannon CP, Neal B, Heerspink HJL. et al. Canagliflozin and Cardiovascular and Renal Outcomes in Type 2 Diabetes Mellitus and Chronic Kidney Disease in Primary and Secondary Cardiovascular Prevention Groups. Circulation. 2019;140:739-50
13. Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N. et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N Engl J Med. 2017;377:644-57
14. Heerspink HJ, Perkins BA, Fitchett DH, Husain M, Cherney DZ. Sodium Glucose Cotransporter 2 Inhibitors in the Treatment of Diabetes Mellitus: Cardiovascular and Kidney Effects, Potential Mechanisms, and Clinical Applications. Circulation. 2016;134:752-72
15. Saggese P, Pandey A, Alcaraz M Jr, Fung E, Hall A, Yanagawa J. et al. Glucose Deprivation Promotes Pseudohypoxia and Dedifferentiation in Lung Adenocarcinoma. Cancer Res. 2024;84:305-27
16. Luo J, Hendryx M, Dong Y. Sodium-glucose cotransporter 2 (SGLT2) inhibitors and non-small cell lung cancer survival. Br J Cancer. 2023;128:1541-7
17. Pandey A, Alcaraz M Jr, Saggese P, Soto A, Gomez E, Jaldu S. et al. Exploring the Role of SGLT2 Inhibitors in Cancer: Mechanisms of Action and Therapeutic Opportunities. Cancers (Basel). 2025 17
18. Basak D, Gamez D, Deb S. SGLT2 Inhibitors as Potential Anticancer Agents. Biomedicines. 2023 11
19. Scafoglio CR, Villegas B, Abdelhady G, Bailey ST, Liu J, Shirali AS. et al. Sodium-glucose transporter 2 is a diagnostic and therapeutic target for early-stage lung adenocarcinoma. Sci Transl Med. 2018 10
20. Canning P, Sorrell FJ, Bullock AN. Structural basis of Keap1 interactions with Nrf2. Free Radic Biol Med. 2015;88:101-7
21. Lo SC, Li X, Henzl MT, Beamer LJ, Hannink M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. Embo j. 2006;25:3605-17
22. Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25:162-71
23. Tian H, Zhang B, Di J, Jiang G, Chen F, Li H. et al. Keap1: one stone kills three birds Nrf2, IKKβ and Bcl-2/Bcl-xL. Cancer Lett. 2012;325:26-34
24. Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543-50
25. Islam SS, Qassem K, Islam S, Parag RR, Rahman MZ, Farhat WA. et al. Genetic alterations of Keap1 confers chemotherapeutic resistance through functional activation of Nrf2 and Notch pathway in head and neck squamous cell carcinoma. Cell Death Dis. 2022;13:696
26. Ren D, Villeneuve NF, Jiang T, Wu T, Lau A, Toppin HA. et al. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci U S A. 2011;108:1433-8
27. Xie J, Lai Z, Zheng X, Liao H, Xian Y, Li Q. et al. Apoptotic activities of brusatol in human non-small cell lung cancer cells: Involvement of ROS-mediated mitochondrial-dependent pathway and inhibition of Nrf2-mediated antioxidant response. Toxicology. 2021;451:152680
28. Xiang Y, Ye W, Huang C, Lou B, Zhang J, Yu D. et al. Brusatol inhibits growth and induces apoptosis in pancreatic cancer cells via JNK/p38 MAPK/NF-κb/Stat3/Bcl-2 signaling pathway. Biochem Biophys Res Commun. 2017;487:820-6
29. Wang T, Chen Z, Chen H, Yu X, Wang L, Liu X. Brusatol inhibits the growth of renal cell carcinoma by regulating the PTEN/PI3K/AKT pathway. J Ethnopharmacol. 2022;288:115020
30. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27-55
31. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440-6
32. Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD. et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76-86
33. Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y. et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99:11908-13
34. Luo J, Sun P, Zhang X, Lin G, Xin Q, Niu Y. et al. Canagliflozin Modulates Hypoxia-Induced Metastasis, Angiogenesis and Glycolysis by Decreasing HIF-1α Protein Synthesis via AKT/mTOR Pathway. Int J Mol Sci. 2021 22
35. Zhou H, Li XM, Meinkoth J, Pittman RN. Akt regulates cell survival and apoptosis at a postmitochondrial level. J Cell Biol. 2000;151:483-94
36. Nitulescu GM, Van De Venter M, Nitulescu G, Ungurianu A, Juzenas P, Peng Q. et al. The Akt pathway in oncology therapy and beyond (Review). Int J Oncol. 2018;53:2319-31
37. Sitthideatphaiboon P, Galan-Cobo A, Negrao MV, Qu X, Poteete A, Zhang F. et al. STK11/LKB1 Mutations in NSCLC Are Associated with KEAP1/NRF2-Dependent Radiotherapy Resistance Targetable by Glutaminase Inhibition. Clin Cancer Res. 2021;27:1720-33
38. Di Federico A, De Giglio A, Parisi C, Gelsomino F. STK11/LKB1 and KEAP1 mutations in non-small cell lung cancer: Prognostic rather than predictive? Eur J Cancer. 2021;157:108-13
39. Secker PF, Beneke S, Schlichenmaier N, Delp J, Gutbier S, Leist M. et al. Canagliflozin mediated dual inhibition of mitochondrial glutamate dehydrogenase and complex I: an off-target adverse effect. Cell Death Dis. 2018;9:226
40. DeBlasi JM, DeNicola GM. Dissecting the Crosstalk between NRF2 Signaling and Metabolic Processes in Cancer. Cancers (Basel). 2020 12
41. Hammad A, Namani A, Elshaer M, Wang XJ, Tang X. "NRF2 addiction" in lung cancer cells and its impact on cancer therapy. Cancer Lett. 2019;467:40-9
42. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029-33
43. Bose S, Zhang C, Le A. Glucose Metabolism in Cancer: The Warburg Effect and Beyond. Adv Exp Med Biol. 2021;1311:3-15
44. Lobo C, Ruiz-Bellido MA, Aledo JC, Márquez J, Núñez De Castro I, Alonso FJ. Inhibition of glutaminase expression by antisense mRNA decreases growth and tumourigenicity of tumour cells. Biochem J. 2000 348 Pt 2: 257-61
45. Turowski GA, Rashid Z, Hong F, Madri JA, Basson MD. Glutamine modulates phenotype and stimulates proliferation in human colon cancer cell lines. Cancer Res. 1994;54:5974-80
46. Galan-Cobo A, Sitthideatphaiboon P, Qu X, Poteete A, Pisegna MA, Tong P. et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res. 2019;79:3251-67
Author contact
![]() Corresponding author: hmshenedu.mo (H.M.S).
Corresponding author: hmshenedu.mo (H.M.S).