Impact Factor ISSN: 1449-2288
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. PCOS and mitochondria...
3. Ferroptosis and mitochondrial...
4. The inflammasome and...
5. ER stress and mitochondrial...
6. Conclusion and future...
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(9):4938-4955. doi:10.7150/ijbs.128537 This issue Cite
Review
Mitochondria and polycystic ovary syndrome: The role of ferroptosis, inflammasomes, and endoplasmic reticulum stress
Xinyi Zhang1,2#, Tong Sun3#, Xinxin Wang4, Yujiu Ma1,2, Liu Cao5* ![]() , Jichun Tan1,2*
, Jichun Tan1,2* ![]()
1. Centre of Reproductive Medicine, Department of Obstetrics and Gynaecology, Shengjing Hospital of China Medical University, Shenyang, Liaoning, China.
2. Key Laboratory of Reproductive Dysfunction Disease and Fertility Remodelling of Liaoning Province, Shenyang, Liaoning, China.
3. Department of Pediatrics, Shengjing Hospital of China Medical University, Shenyang, Liaoning, China.
4. Department of Reproductive Medicine, Shenyang Maternity and Child Health Hospital, Shenyang, Liaoning, China.
5. Clinical Translational Research Center, Shengjing Hospital of China Medical University, Shenyang, Liaoning, China.
*Jichun Tan and Liu Cao contributed equally.
#Xinyi Zhang and Tong Sun contributed equally.
Received 2025-11-17; Accepted 2026-4-9; Published 2026-5-1
Abstract

Polycystic ovary syndrome (PCOS) poses a major threat to women of reproductive age and is strongly associated with metabolic and inflammatory abnormalities. Over the past decade, tremendous progress has been made in our understanding of signaling events regulated by mitochondria. Emerging evidence underscores mitochondrial dysfunction as a central pathophysiological hub in PCOS. The intricate crosstalk among mitochondrial dysfunction, ferroptosis, inflammasomes, and endoplasmic reticulum (ER) stress creates a pathological network that underpins ovarian dysfunction, metabolic abnormalities, and chronic inflammation in PCOS, highlighting promising novel targets for diagnosis and therapeutic intervention in this complex disorder.
Keywords: mitochondria, ferroptosis, inflammasome, endoplasmic reticulum stress, polycystic ovary syndrome
1. Introduction
Polycystic ovary syndrome (PCOS) is the most common reproductive and endocrine disorder affecting between 10% and 13% of women, based on the population under investigation and the diagnostic standards used [1, 2]. Its primary characteristics include chronic ovulatory dysfunction, hyperandrogenism (HA), and polycystic ovarian morphology, with clinical manifestations such as menstrual irregularities, acne, hirsutism, and infertility [3]. Importantly, PCOS is not merely a reproductive disorder. The majority of women with PCOS exhibit metabolic disturbances, including insulin resistance (IR), hyperinsulinemia, dyslipidemia, overweight/obesity, impaired glucose tolerance, and an elevated risk of type 2 diabetes [4-6]. The pathogenesis of PCOS is complex and influenced by genetic, environmental, and hormonal factors. PCOS is associated with a higher risk of adverse pregnancy outcomes, including miscarriage, gestational diabetes, and hypertensive disorders of pregnancy [7, 8]. Later in life, women with PCOS face significantly elevated risks of cardiovascular disease and endometrial cancer [9, 10]. Given the complexity of PCOS and its lifelong health implications, current treatment mainly relies on symptom-based management and long-term health monitoring [11, 12].
Early diagnosis and treatment of PCOS continue to pose challenges. Several potential biomarkers of PCOS have been reported, spanning hormonal, metabolic, oxidative stress, inflammatory, and microRNA-related categories [13]. Nevertheless, due to the heterogeneity of the disease, emerging evidence suggests that biomarker signatures may vary across PCOS phenotypes, and many women experience delayed diagnosis [14]. Lifestyle modification remains the cornerstone of PCOS management. In addition, oral contraceptives are commonly used for menstrual irregularities and hyperandrogenism. Insulin sensitizers such as metformin are used to manage metabolic disturbances in PCOS. For infertility, letrozole is recommended as the first-line ovulation induction agent, with clomiphene citrate, gonadotrophins, and laparoscopic ovarian surgery as alternative options [15, 16]. Nevertheless, there remains a dearth of more precise and effective therapeutic agents. Thus, elucidating the pathogenesis of PCOS is crucial for reducing its incidence and improving clinical outcomes.
Mitochondria, as key regulators of cellular metabolism, proliferation, aging, and programmed cell death, have emerged as a focal point in PCOS pathogenesis research, attracting increasing attention in recent years [17, 18]. Furthermore, mitochondria can influence the incidence and progression of PCOS by modulating ferroptosis, inflammasome activation, and ER stress via multiple pathways, including reactive oxygen species (ROS) generation and alterations in mitochondrial metabolism [19-21]. The above findings have established the role of mitochondria in PCOS. Therefore, we integrate the current literature on the complex interplay between mitochondrial damage, ferroptosis, inflammasomes, and ER stress in the molecular pathogenesis of PCOS.
2. PCOS and mitochondria pathophysiology
2.1 Mitochondrial ROS
The mainly documented mitochondrial dysfunction in PCOS pathogenesis is the elevated production of mitochondrial ROS. Mitochondria are the main sources of intracellular ROS. During normal aerobic respiration, the electron transport chain (ETC) transfers electrons to molecular oxygen for water production. However, a minor proportion of electrons leak directly to oxygen, forming superoxide anions (O2-), a type of ROS. O2- can be further transformed by superoxide dismutase (SOD) into hydrogen peroxide (H2O2); H2O2 is relatively stable, has high membrane permeability, and can diffuse within the cell and participate in signal transduction [22]. ROS are involved in the modulation of various molecules and signaling pathways, including the activation of protein kinase signaling cascades, the regulation of transcription factor activity, and calcium ion homeostasis [23, 24]. Under specific pathological conditions such as mitochondrial dysfunction, hypoxia, or inflammation, pronounced electron leakage occurs within the ETC, leading to substantial ROS generation. Moreover, ROS production represents a critical mechanism of immune defense that promotes the release of chemokines and cytokines from immune cells, thereby enhancing the immune response [25].
HA and IR are the primary pathophysiological mechanisms of PCOS. Furthermore, HA elevates the risk of IR, and both are closely associated with the onset of oxidative stress, which are significant factors contributing to oxidative stress in PCOS. Research has demonstrated that PCOS patients with the HA phenotype have more severely impaired antioxidant function [26]. In these patients, HA induces the expression of ovarian aldose reductase, leading to an increase in the flux of the polyol pathway, and also increases ovarian lipid peroxidation, reduces catalase activity and glutathione (GSH) content, further damaging ovarian function [27]. Lower mitochondrial efficiency and increased level of mitochondrial H2O2 emissions in obese women with IR compared with lean women have been reported in a clinical randomized controlled trial. Furthermore, the level of mitochondrial H2O2 emissions in the obese group decreased after exercise [28]. Similarly, the production of ROS increases in women with PCOS, accompanied by a reduction in GSH levels and oxygen consumption, and may be alleviated by the administration of metformin, a natural antioxidant [29, 30]. Excessive oxidation reactions and the weakening of antioxidant capacity may be key factors that accelerate the onset of PCOS. An animal study indicated that pregnant PCOS rats exhibited a mismatch between oxidative and antioxidative stress reactions in the gravid uterus [31]. Results of a study by Zhang et al. also support this conclusion, reporting that the mitochondria-ROS-SOD1/Nrf2 pathway in the placenta of PCOS rats is directly linked to the detrimental effects of IR and HA on embryonic survival. SOD1 is an important protein involved in oxidative stress, while Nrf2 participates in antioxidant responses [32]. Nrf2-Foxo1-ROS and ROS/p38/JNK pathways have also been reported in PCOS models [33, 34]. Oxidative stress, as a key pathological response of the body to metabolic imbalance, exhibits a significantly abnormal activation state in the ovarian tissues of PCOS patients. Its interaction with IR and HA may may form a vicious cycle that drives disease progression. In addition to direct damage, ROS also plays a significant role in mitochondrial metabolism and quality control processes [35].
2.2 Mitochondrial metabolism
Mitochondria serve as energy-producing organelles [36]. The tricarboxylic acid (TCA) cycle and oxidative phosphorylation occur within the mitochondria and produce adenosine triphosphate (ATP). Mitochondria also act as central intermediaries connecting glucose, fatty acid, and amino acid metabolism. Pyruvate is converted into acetyl-CoA in mitochondria to participate in the TCA cycle, while β-oxidation breaks down fatty acids into acetyl-CoA, thereby supporting the TCA cycle and energy generation. Metabolic disturbances can result in the reprogramming of mitochondrial energy metabolism [37]. PCOS has been found to be associated with abnormalities in mitochondrial energy metabolism, with studies mainly focusing on ovarian granulosa cells and showing decreased ATP production and reduced glycolysis levels [38, 39]. Increased levels of ROS and lower expression of glycolysis-associated genes, including glucose transporter-1 (GLUT1), phosphofructokinase (PFK), and lactate dehydrogenase A (LDHA), have been reported in granulosa cells of PCOS women compared with non-PCOS women, indicating that the low oocyte competence in PCOS may be associated with mitochondrial dysfunction and abnormal glycolysis [40]. A clinical study by Mazloomi et al. also found abnormalities in ATP content and glucose metabolism disorders in granulosa cells of PCOS patients. Women with PCOS show lower ATP concentrations and decreased expression levels of key glycolytic enzymes, including PFK and hexokinase 1 (HK1), accompanied by a lower glycolysis rate compared with a control group [41]. In addition to reduced ATP synthesis, increased mitochondrial ROS, and decreased glycolysis, Zhang et al. verified insufficient mitochondrial oxidative phosphorylation levels in granulosa cells of PCOS women. These changes were found to be regulated by Sirtuin 3 (SIRT3) signaling in KGN cells [42]. Cao et al. found that follicular fluid-derived exosomal miR-155-5p and miR-143-3p participate in the regulation of glycolysis and apoptosis of granulosa cells in the PCOS model [43]. However, in ovarian stromal cells of a PCOS model, glycolysis may actually increase, but this is more related to promoting the inflammation and fibrosis than to directly supporting follicle development [44].
2.3 Mitochondrial quality control
2.3.1 Mitochondrial biogenesis
The maintenance of mitochondrial homeostasis is governed mainly by mitochondrial quality control (MQC), which involves mitochondrial biogenesis, dynamics, and autophagy. The process of mitochondrial biogenesis maintains the quantity of mitochondria and replaces aging or damaged mitochondria with new, healthy ones. This process is rigorously regulated by the peroxisome proliferator-activated receptor gamma coactivator (PGC-1α) through the activation of Nrf1/2 and mitochondrial transcription factor A (TFAM) [45, 46]. The transcription and replication of mitochondrial DNA (mtDNA) are facilitated by signaling molecules such as TFAM, which is subsequently translated into proteins and assembled into functional mitochondria. Mitochondria possess their own genetic material, mtDNA, which encodes various molecules essential for mitochondrial function. Mutations in the mtDNA can lead to mitochondrial dysfunction and diseases [47].
A randomized, triple-blind, placebo-controlled clinical trial revealed that granulosa cells of PCOS women have lower expression of important genes involved in mitochondrial biogenesis, such as TFAM and PGC-1α. This expression level may be increased by resveratrol administration, which activates Sirtuin 1 (SIRT1) and improves the mtDNA copy number [48]. In a dehydroepiandrosterone (DHEA)-induced PCOS mouse model, Safaei et al. found that vitamin D3 improved mitochondrial biogenesis by stimulating the mitogen-activated protein kinase (MAPK) pathway in granulosa cells [49]. Furthermore, Pang et al. reported the role of SIRT3/Foxo1/PGC-1α in KGN cells of a PCOS model, and Sun et al. showed that mitochondrial biogenesis could also be regulated by circadian clock genes REV-ERBs [50, 51]. Reduced mitochondrial biosynthesis leads to decreased activity of antioxidant enzymes (such as SOD and GPX), resulting in the ROS accumulation [52]. Excessive ROS further damages mtDNA and membrane structures, ultimately accelerating follicular closure [53].
mtDNA copy number and variants are also important factors in PCOS. According to Tharayil et al., women with PCOS had lower mtDNA copy numbers and also had mtDNA variations. PCOS is strongly associated with mtDNA variations such as 1488T, 9670G, 12556G, 3308G, 9200G, 15914T, 14480G, and 5426G [54]. In addition, a few studies have reported on mitochondrial haplogroups and population genetics related to PCOS susceptibility. Mitochondrial haplogroups are genetic lineages classified based on variations in mtDNA sequences, reflecting the evolutionary history of human maternal inheritance. Recent reports have indicated that specific mitochondrial haplogroups may serve as genetic backgrounds, interacting with functional mutations to participate in the pathogenesis of PCOS. Leng et al. reported a case of a Chinese PCOS patient. The researchers conducted whole mitochondrial genome sequencing on this patient and found that she carried a set of polymorphic variations specific to mitochondrial haplogroup F2. Additionally, the patient had two homoplasmy point mutations: the ND5 T12338C mutation and the tRNASer (UCN) C7492T mutation. As the genetic background of the patient, haplogroup F2 itself may not directly cause disease, but when combined with the above two functional mutations, it may synergistically lead to mitochondrial dysfunction and increase the risk of PCOS [55]. Another study by the same group reported a three-generation family with a maternally inherited pattern. Multiple members of this family suffered from metabolic syndrome, and a female in the third generation also presented with PCOS. Mitochondrial gene analysis of the family members revealed that all affected individuals carried a set of genetic variations belonging to the East Asian mitochondrial haplogroup B4b1c. Moreover, the researchers discovered three homoplasmy mitochondrial tRNA mutations, which led to significant mitochondrial dysfunction [56]. mtDNA is maternally inherited in most animals and is characterized by a rapid evolutionary rate and haploid inheritance, making it an important tool in population genetics research. The genetic variations, distribution, dynamics and evolutionary significance of mtDNA within a population are the main contents of mitochondrial population genetics. Based on the analysis of mtDNA haplotypes, it has been widely used to assess the genetic structure, gene flow, historical dynamics and systematic geographical information of populations [57]. In a study of the Chinese population, Chen et al. identified that certain mtDNA D-loop mutations (G207A, 16036GGins, 16049Gins) and haplotype A15 confer protection against PCOS, and further demonstrated that PCOS patients have significantly higher mtDNA copy numbers than controls [58]. A recent cross-ethnic study by Nawaz et al. analyzed the mitochondrial transfer RNA (mt-tRNA) genes in 64 Pakistani patients with PCOS and compared the identified mutations with patients from other ethnic groups. Eight variants in five mt-tRNA genes were found, most of which were novel and occurred in highly conserved nucleotides of tRNA. The study revealed that certain mt-tRNA genes carrying PCOS-associated mutations may be specific to particular ethnic populations [59].
2.3.2 Mitochondrial dynamics
Mitochondria are extremely dynamic organelles that undergo constant cycles of fission and fusion, changing their morphology, size, and distribution. This process, known as mitochondrial dynamics, is crucial for maintaining cellular homeostasis. Fusion involves the merging process of both the inner and outer mitochondrial membranes, enhancing the resilience of the mitochondrial network and facilitating the exchange of mitochondrial contents. The inner mitochondrial membrane fusion protein optic atrophy 1 (OPA1) and the outer mitochondrial membrane fusion proteins mitofusin 1 and 2 (MFN1/2) co-regulate this process [60, 61]. Conversely, mitochondrial fission partitions the tubular mitochondrial network into smaller fragments, which aid in the removal of depolarized mitochondria via mitophagy. The process of fission is primarily regulated by dynamin-related protein 1 (DRP1). After phosphorylation-induced activation, DRP1 translocates to the outer mitochondrial membrane and interacts with mitochondrial fission factor (MFF) to drive the fission process. Under normal physiological conditions, mitochondrial fusion and fission are tightly balanced to maintain mitochondrial integrity. Disruption of this equilibrium impairs mitochondrial function and contributes to disease [62].
To date, there have been few studies on dynamic mitochondrial changes in PCOS. Salehi et al. found that mitochondrial fission increased and protein levels of DRP1 and phospho-DRP1 (Ser616) were upregulated in a dihydrotestosterone (DHT)-induced rat model of PCOS [63]. Li et al. also reported that the fission cofactors Fis1 and MFF were significantly increased in this model. Although DRP1 expression level did not change, activated phospho-DRP1 (Ser616) increased markedly, whereas the inactivated phospho-DRP1 (Ser637) protein expression level decreased significantly. The addition of the DRP1 inhibitor Mdivi-1 has the potential to reverse this process. The imbalance in DRP1 phosphorylation might have been involved in the mechanism underlying increased mitochondrial fission [64]. Another study has reported the effect of mitochondrial fusion on PCOS and found that MFN2 protein expression levels were decreased in oocytes and granulosa cells of a PCOS model [21]. However, a clinical study has demonstrated that in ovarian granulosa cells of patients with hyperandrogenic PCOS, the mRNA expression of mitochondrial fusion genes mitoguardin 1 and 2 was significantly upregulated, with expression levels positively correlated with serum testosterone concentrations [65]. HA may enhance compensatory mitochondrial fusion, but evidence from animal models indicates that prolonged androgen exposure may ultimately lead to a predominance of fission and impairment of the fusion process. The reduction in mitochondrial fusion and the concomitant increase in mitochondrial fission exacerbate mitochondrial dysfunction, thereby promoting elevated levels of mitophagy [66].
2.3.3 Mitophagy
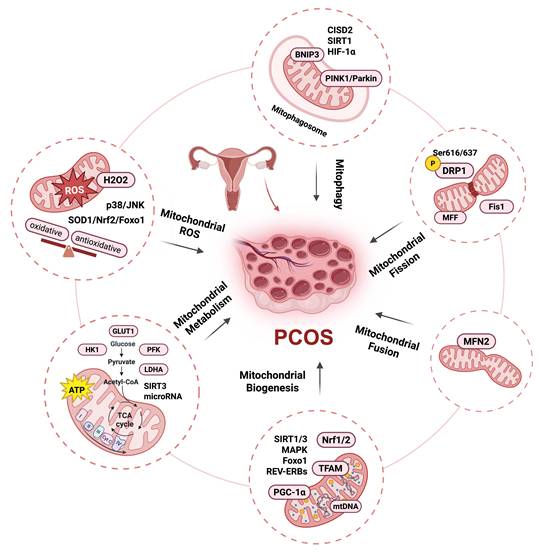
Mitophagy is a selective cellular process by which damaged or dysfunctional mitochondria are removed. This process involves the sequestration of mitochondria by autophagosomes and their subsequent fusion with lysosomes, and is primarily mediated by the PINK1-Parkin pathway. Mitophagy can also occur via non-ubiquitination-dependent pathways, such as the BCL2/adenovirus E1B interacting protein 3-like (BNIP3L) or FUN14 domain containing 1 (FUNDC1) pathway [67, 68]. Current research indicates that mitophagy in PCOS does not simply exhibit unidirectional enhancement or reduction but rather exists in a state of imbalance. This imbalance is a crucial factor contributing to mitochondrial dysfunction in granulosa cells and ovarian tissues. Mitophagy-associated proteins PTEN-induced kinase 1 (PINK) and Parkin (an E3 ubiquitin ligase) are significantly upregulated in the granulosa cells of PCOS patients and can be regulated by the CDGSH iron sulfur domain 2 (CISD2) pathway. CISD2 activation can inhibit mitophagy via the PINK/Parkin pathway and the binding of mitochondria to lysosomes [69]. Yi et al. also showed that Parkin and PINK1 protein levels were upregulated and that this upregulation could be alleviated by the addition of melatonin via activation of the SIRT1 pathway [70]. However, some studies have demonstrated that mitophagy is impeded in PCOS. In the granulosa cells of patients with PCOS and in letrozole-induced rat models, the HIF-1α/BNIP3 mediated mitophagy is suppressed, resulting in a decrease in the clearance capacity of damaged mitochondria and accumulation of ROS, thereby inducing mitochondrial dysfunction [71]. Evidence from studies on PCOS demonstrate the role of mitochondria as a signaling hub in PCOS, as illustrated in Figure 1.
Mitochondria as a signaling hub in PCOS. Mitochondrial damage is primarily associated with mitochondrial oxidative stress, metabolic dysfunction, and quality control disorders. Clinical and basic studies show that PCOS exhibits an oxidative and antioxidative stress responses imbalance: the level of ROS and H2O2 emissions increase, accompanied by a decrease in oxygen consumption and GSH levels, which may be alleviated by metformin. Mitochondria-ROS-SOD1/Nrf2, Nrf2-Foxo1-ROS, and ROS/p38/JNK pathways have been reported in PCOS models. PCOS patients also exhibit reduced ATP content and decreased glycolysis that could by regulated by follicular fluid-derived exosomal miR-143-3p/miR-155-5p; glycolysis-associated gene level downregulation including GLUT1, LDHA, PFK, and HK1; and decreased mitochondrial oxidative phosphorylation levels regulated by SIRT3. Mitochondrial quality control disorder in PCOS including mitochondrial biogenesis decreases regulation by PGC-1α, TFAM, SIRT1/3, Foxo1, REV-ERBs, and MAPK; mtDNA copy number decreases; mitochondrial fission increase regulated by DRP1, p-DRP1 (Ser616/637), Fis1, and MFF; mitochondrial fusion decrease regulated by MFN2; mitophagy increase regulated by PINK/Parkin pathway, CISD2, and SIRT1, and decrease regulated by HIF-1α/BNIP3 pathway. This figure was created by Biorender (https://biorender.com/).
3. Ferroptosis and mitochondrial dysfunction in PCOS
3.1 Ferroptosis and ovarian follicle development
In 2012, Stockwell discovered ferroptosis, a type of programmed cell death caused by iron-dependent lipid peroxidation. Unlike apoptosis and other types of cell death, ferroptosis is characterized by shrunken and dense mitochondria, rounding and swelling of cells, cell rupture with intact nuclei, and chromatin condensation [72]. Ferroptosis is regulated by various cellular metabolic processes, including redox equilibrium, iron homeostasis, mitochondrial function, and glucose, lipid, and amino acid metabolism. Interconnected processes such as lipid metabolism, ROS biology, and iron control contribute to its initiation and progression [73]. ROS produced by mitochondria lead to lipid peroxidation, resulting in irreparable damage to the cell membrane and ferroptosis.
Iron plays an important role in regulating ferroptosis sensitivity. The Fenton reaction generates free radicals that react with phospholipid-containing polyunsaturated fatty acid (PUFA-PL) chains, resulting in the formation of phospholipid hydroperoxides (PLOOHs), a hallmark of ferroptosis [74]. Glutathione peroxidase 4 (GPX4) is the most important enzyme for suppressing ferroptosis. The system XC-activation, a cystine/glutamate antiporter for amino acid transport, causes extracellular cystine to be carried within the cell, intracellular glutamate is transported outside the cell. Glutathione, produced by cysteine, converts PLOOHs to phospholipid alcohols (PLOHs) via stimulating GPX4 to inhibit cell ferroptosis [75]. Ferroptosis has been indicated in various pathophysiological processes such as immunity, aging, cardiovascular diseases, neurodegenerative diseases, endocrine metabolic diseases, and cancer [76].
Studies have shown that ferroptosis is related to the growth and development of ovarian follicles. A minor quantity of iron is innocuous to the ovaries, however, excessive iron can impair ovarian hormone production and follicle development. This significantly affects the endocrine system and fertility of female mice, potentially leading to infertility. Moreover, the ovarian reserve function of their offspring may also be genetically influenced [77]. Ferritin can up-regulate the expression of NF-κB and inducible nitric oxide synthase (iNOS) in rat ovaries and down-regulate the expression of GPX4, leading to a decrease in estrogen levels. Excessive iron can also cause iron-dependent inflammatory and oxidative stress responses, resulting in ovarian damage and dysfunction [78]. Under conditions of obesity, ferroptosis can be activated and contribute to the depletion of primordial follicles. Studies have shown that during the primordial to primary transition stage of follicles in obese mice, ferroptosis and cellular oxygen-related signaling pathways are significantly enriched. In the ovaries of obese mice, the depletion of primordial follicles increases, fat deposition around follicles is higher, and granulosa cell proliferation is enhanced [79]. Ferroptosis also assumes a significant role in the process of ovarian aging. Research has indicated that in granulosa cells of patients with diminished ovarian reserve and advanced age, GPX4 expression is downregulated and GSH levels are decreased, suggesting that ferroptosis participates in the process of ovarian aging. In vitro experiments have revealed that ferroptosis inducers impede granulosa cell growth by downregulating GPX4, whereas ferroptosis inhibitors can upregulate GPX4 expression and reverse this inhibitory effect. In mouse models, GPX4 expression is reduced in the oocytes of aged mice, and treatment with ferrostatin-1 (Fer-1) can enhance the number and quality of retrieved oocytes [80].
3.2 Ferroptosis and PCOS
Kim et al. found that metabolic abnormalities such as hepatocellular adenoma with iron-related disease (HAIR) are associated with elevated circulating iron levels [81]. Higher serum ferritin levels have also been reported in PCOS women, indicating iron overload [82]. Zhang et al. reported an association between ferroptosis and PCOS [83]. In their study, maternal rats treated with 5α-DHT and insulin exhibited reduced levels of GPX4 and glutathione, as well as upregulated levels of malondialdehyde and glutathione + glutathione disulfide in the gravid uterus, and these compounds may participate in regulating ferroptosis. They also observed aberrant expression of ferroptosis-associated genes including Acsl4, Slc7a11, Tfrc, and Gclc, as well as increased iron deposition. Ferroptosis is mediated by the MAPK signaling system, which is triggered in the gravid uterus and includes extracellular signal-regulated kinase (ERK), p38, and c-Jun NH2-terminal kinase (JNK).
In addition to ferroptosis in the uterus, Hu et al. observed placental ferroptosis in a PCOS model [84]. After administration of the antioxidant N-acetylcysteine, GPX4 protein levels increased, and ferroptosis in the gravid uterus and placenta was reversed. Administration of N-acetylcysteine could be a viable therapeutic approach for PCOS. Granulosa cells of the ovary also undergo ferroptosis in patients with PCOS and may be regulated by the circRHBG/miR-515/SLC7A11 axis [85]. Tan et al. reported that miR-93-5p could also regulate ferroptosis in granulosa cells by regulating the NF-κB signaling pathway [86]. Additionally, a recent study discovered that ferroptosis was elevated in PCOS women ovaries and in rat models induced by DHEA. The addition of Fer-1, a ferroptosis inhibitor, was found to alleviate a cluster of PCOS traits, including HA and ovulatory dysfunction. HA is regarded as a key factor triggering ferroptosis. In granulosa cells of PCOS women and ovaries of PCOS rats, this study found that concentrations of Fe2+ and malondialdehyde were increased, protein levels of nuclear receptor coactivator 4 (NCOA4) were upregulated, and GPX4 and ferritin heavy chain 1 (FTH1) protein levels were downregulated [87]. Thus, activation of NOCA4-dependent ferritinophagy may be significant processes underlying ferroptosis in ovarian granulosa cells. Bioinformatics analyses revealed 14 differentially expressed genes in granulosa cells between PCOS patients and non-PCOS women, including LPIN1, BNIP3, DDIT4, ATF3, NOS2, NQO1, SLC2A6, and SLC2A1, which were enriched in mitochondrial outer membrane, ROS metabolic processes, and antioxidant activity, as ferroptosis [88]. Although current evidence consistently indicates elevated levels of ferroptosis in PCOS, heterogeneity across study populations may exist, and these findings do not imply a universal increase in all individuals with PCOS.
3.3 Ferroptosis regulated by mitochondria in PCOS
Mitochondria play a key role in regulating ferroptosis [89, 90]. Ferroptosis causes dramatic morphological changes in the mitochondria, including mitochondrial shrinkage, fragmentation, and cristae enlargement [91]. Zhang et al. observed shrunken mitochondria with electron-dense cristae and increased levels of the mitochondria-encoded gene Dpp4, which can induce ferroptosis in the uteri of rats [83]. The most significant mechanism of ferroptosis in mitochondria is the production of ROS. Mitochondrial ROS production induces ferroptosis by promoting lipid peroxidation [92]. An in vitro study discovered the role of ROS signaling in ferroptosis triggered by ferric ammonium citrate (FAC) in KGN cells. FAC promotes the expression of NADPH oxidase 1 (NOX1) signaling, which is a vital regulator of oxidative stress. In this work, inhibition of NOX1 reversed the effects of FAC, increased GPX4 levels, and reduced Fe2+ release. This study also found that the transferrin receptor can increase iron level, promote ROS release, increase ACSL4 levels, activate mitophagy via the PINK1 pathway, and induce lipid peroxidation, indicating the role of the NOX1/PINK1/ACSL4 pathway in PCOS [93]. Li et al. also reported the role of ROS in PCOS ferroptosis. Moreover, they found that treatment with baicalein, a flavonoid compound from the Lamiaceae family, could inhibit oxidative stress and ameliorate PCOS by suppressing chronic inflammation and lipid peroxidation and by modulating mitochondrial functions through regulation of glutathione peroxidase and the FTH1 signaling pathway to restrain ferroptosis in KGN cells [94]. Another recently published study reported that n-3 PUFA dramatically reduces mitochondrial function in PCOS granulosa cells, resulting in a shift from high to low mitochondrial membrane potential. n-3 PUFA may also lead to mitochondrial sequestration and enhance the density of bilayer structures in KGN cells, which is correlated with the ultrastructure of ferroptosis [95].
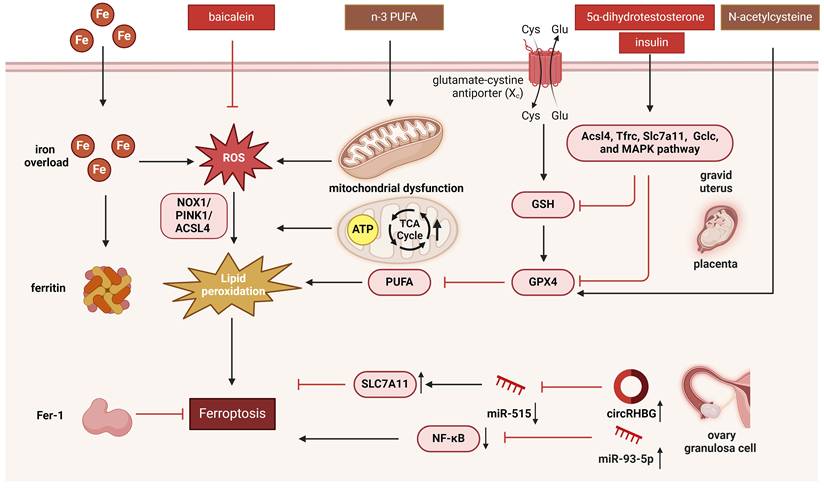
Mitochondria are the primary organelles responsible for ATP production. AMP-activated protein kinase could inhibit the synthesis of certain PUFAs and ferroptosis by phosphorylating and suppressing acetyl-CoA carboxylases [96]. Mitochondria also perform biosynthetic functions during cellular metabolism. The TCA cycle and several anaplerotic processes that repel the TCA cycle, such as glutaminolysis, are located in the mitochondria. Inhibition of glutaminolysis or glutamine deficiency significantly reduces ferroptosis. The mechanisms underlying the regulation of ferroptosis by the TCA cycle are likely related to electron transport and fatty acid production [97]. Ferroptosis can also be influenced by other mitochondrial pathways, including those involved in sulfur transfer, p53, Nrf2-regulated iron and lipid metabolism, and the mitochondrial voltage-dependent anion channel pathways [98, 99]. Changes in the regulation of mitochondrial dynamics also affect mitochondrial function and consequently induce ferroptosis [100]. Collectively, these findings establish mitochondria as central hubs in the regulation of ferroptosis. On one hand, mitochondrial damage serves as both a trigger and a consequence of ferroptotic cell death. On the other hand, key ferroptosis regulators are intrinsically linked to mitochondrial metabolism. Notably, interventions such as baicalein and n-3 PUFA appear to ameliorate PCOS phenotypes by restoring mitochondrial function and inhibiting ferroptosis [101]. The regulation of mitochondria and the underlying mechanisms remain incompletely understood and warrant further investigation. The mechanism of mitochondrial regulation of ferroptosis in PCOS is presented in Figure 2.
The mechanism of ferroptosis regulated by mitochondria in PCOS. Ferroptosis is characterized by shrunken and dense mitochondria driven by iron-dependent lipid peroxidation, and can be regulated by mitochondria. Ferroptosis in PCOS can be regulated by mitochondria via mitochondrial ROS regulated by NOX1, glutathione peroxidase, and FTH1; mitochondria-encoded gene Dpp4; mitophagy regulated by the NOX1/PINK1/ACSL4 pathway; TCA cycle and ATP production; p53 pathway and Nrf2-regulated iron and lipid metabolism pathways; and the mitochondrial voltage-dependent anion channel pathway. This figure was created by Biorender (https://biorender.com/).
4. The inflammasome and mitochondrial dysfunction in PCOS
4.1 NLRP3 inflammasome and PCOS
PCOS is associated with a chronic inflammatory status [102]. Under pathological conditions, oocyte development may be disturbed, resulting in follicular atresia and ovulatory dysfunction [103]. Elevated levels of pro-inflammatory cytokines, C-reactive protein, and white blood cells have been reported in patients [104]. Recently, inflammasomes have been demonstrated to be implicated in the pathophysiology of numerous inflammatory and metabolic illnesses [105].
As inflammasome-related cytokines, IL-18 and IL-1β are involved in the ovulatory process and follicular dynamics [106]. Moreover, increased levels of IL-18 and IL-1β have been reported in the follicular microenvironment of PCOS women [107]. Inflammasomes are intracellular multimeric complexes that recognize pathogen-associated and damage-associated molecular patterns, as first reported in 2002 [108, 109]. Inflammasome assembly induces the autoproteolytic processing of caspase-1, prompting the cleavage of its cellular substrates. Active caspase-1 cleaves the precursors of IL-18 and IL-1β to aid their maturation [110-113]. Additionally, activated caspase-1 cleaves gasdermin D (GSDMD) into activated N-GSDMD, leading to cell membrane pore formation and cellular lysis, thereby inducing pyroptotic cell death and cytokine release, which are associated with various physiological processes [111, 114, 115]. To elucidate the role of inflammasomes in the follicular microenvironment, it is crucial to explore the mechanisms underlying PCOS progression.
Several types of inflammasomes have been identified, of which the nod-like receptor (NLR) family pyrin domain-containing 3 (NLRP3) inflammasome is the most studied [116, 117]. Intracellular and extracellular pathogenic signals stimulate the assembly and activation of the inflammasome complex. It has been documented those certain ionic signals, including ATP, lysosomal rupture, ROS, K+ efflux, Ca2+ signaling, and mitochondrial malfunction, activate the NLRP3 inflammasome [118]. Moreover, abnormal metabolic status is related to the activation of NLRP3 inflammasomes. NLRP3 activation is implicated in steroidogenesis, oocyte maturation, autophagy, and apoptosis [119]. HA is a characteristic feature of PCOS. A study found that hyperandrogen levels increase ROS production in ovarian cells by activating NLRP3 in PCOS rats [120]. Higher levels of NLRP3 inflammasomes have been reported in the peripheral blood mononuclear cells and granulosa cells of women with PCOS than in the control group [119, 121]. Liu et al. detailed the addition of formononetin in vivo and in vitro by inhibiting inflammation, apoptosis, and oxidative stress caused by the NLRP3 inflammasome [122]. Li et al. observed that the gut microbial metabolite indole-3-propionic acid (IPA) alleviates PCOS in mice by regulating the aryl hydrocarbon receptor (AhR)/NLRP3 axis [123]. A recent systematic review and meta-analysis also indicated that NLRP3 is upregulated in patients with PCOS and animal models [124]. Several free fatty acid levels are significantly associated with mature IL-18 in follicular fluids, and oleic acid treatment activates inflammasome signaling in KGN cells [125]. In addition, Liu et al. stimulated KGN cells with the follicular fluid from women with PCOS and found that it activated NLRP3 inflammasomes, the NF-κB pathway, and impaired mitochondria structure and function in KGN cells were found [107].
Abnormal metabolic status is associated with the activation of the NLRP3 inflammasome. Metabolic pathway alterations occur during macrophage activation, indicating that different metabolic processes lead to different inflammasome fates, including the regulatory effects of glycolytic enzymes, TCA cycle metabolites, and lipid pathways on inflammasomes. Taken together, the altered follicular microenvironment in PCOS induces inflammatory stress in oocytes and the surrounding granulosa cells. Several studies have demonstrated the involvement of inflammasome-dependent pathways and pyroptosis in ovarian dysfunction. For instance, exposure to the endocrine-disrupting substance di-(2-ethylhexyl) phthalate (DEHP) can cause reproductive system dysfunction. Sun et al. discovered a novel mechanism by which DEHP induces pyroptosis in ovarian granulosa cells via the SLC39A5/NF-κB/NLRP3 axis, thereby impairing ovarian function [126].
4.2 GSDMD-dependent pyroptosis and PCOS
GSDMD-dependent macrophage pyroptosis regulates various physiological processes involved in inflammation [127]. Ovarian granulosa cell survival is essential for PCOS development and progression. Cellular pyroptosis occurs in PCOS and is associated with inflammasome activation. Wang et al. demonstrated that high androgen levels stimulate chronic low-grade inflammation in the ovaries of PCOS mice by activating the NLRP3 inflammasome and induce follicular dysfunction, ovarian granulosa cell pyroptosis, and ovarian interstitial cell fibrosis [128]. Huang et al. reported that GSDMD-dependent macrophage pyroptosis impairs estrogen synthesis and induces apoptosis of granulosa cells in PCOS mice. Additionally, metformin treatment regulated gut microbiota abundance and inhibited macrophage pyroptosis in the ovaries, thus ameliorating PCOS [129].
However, the underlying molecular mechanism remains unclear. Cai et al. discovered that Wilms' tumor 1-associated protein (WTAP), a critical regulator of the RNA N6-methylase complex, is upregulated in granulosa cells, resulting in inflammasome overactivation. Overexpression of WTAP in granulosa cells stabilizes the mRNA expression of the inflammasome component apoptosis-associated speck-like protein (ASC), causing granulosa cell pyroptosis in PCOS [130]. The role of miRNAs in regulating PCOS has been reported. For example, miR-29a-3p is downregulated in PCOS. Histone deacetylase 1 (HDAC1) inhibits granulosa cell pyroptosis in PCOS through deacetylation to regulate the H19/miR-29a-3p/NLRP3 axis [131]. Wu et al. found that Guizhi Fuling Wan intervention upregulated the expression level of miR-29b-3p and downregulated the expression level of H19, thereby inhibiting granulosa cell autophagy in PCOS [132]. Another study found that miR-1224-5p reduces NLRP3 inflammasome activation, IL-1β production, and NF-κB p65 nuclear translocation, lowering inflammation and preventing PCOS development [133]. These findings improve our understanding of PCOS and provide potential new targets for its treatment.
4.3 Inflammasome regulated by mitochondria in PCOS
Mitochondria serve as organizational hubs for the assembly and activation of the NLRP3 inflammasome [134]. Mitochondrial damage is implicated in the induction of inflammasome complexes. ROS is a common signal that activates the NLRP3 inflammasome [135]. Impaired mitochondrial autophagy leads to prolonged mitochondrial ROS production [136]. NLRP3 is directly activated by mitochondria-derived effector molecules. Zhong et al. found that mtDNA synthesis is crucial for NLRP3 signaling. The synthesis of oxidized mtDNA fragments requires mtDNA synthesis dependent on CDGSH iron sulfur domain 2 (CMPK2), a rate-limiting enzyme that provides deoxyribonucleotides for mtDNA synthesis [137]. Oxidized mtDNA, released into the cytosol upon mitochondrial dysfunction, activates the NLRP3 inflammasome [138]. The binding of mtDNA to the NLRP3 inflammasome complex is necessary for this activation.
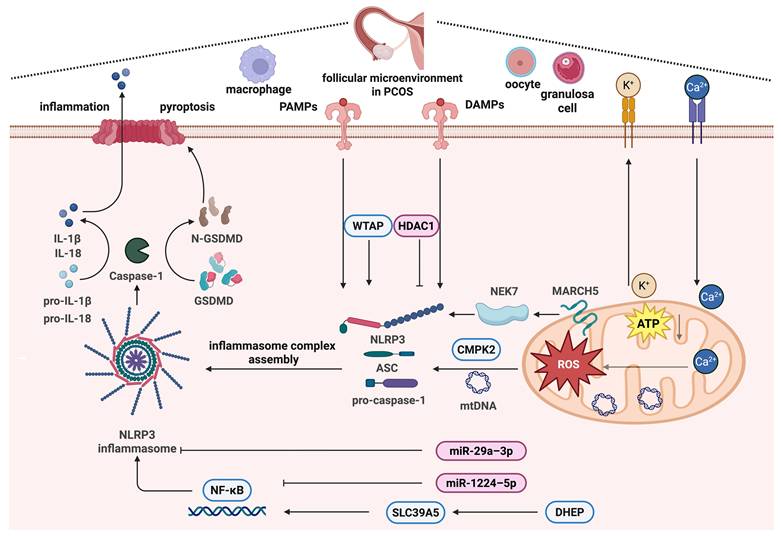
Moreover, mtDNA release depends on inflammasome activation and mitochondrial ROS-dependent mechanisms [139]. Mitochondria play a critical role in the activation of the NLRP3 inflammasome by binding NLRP3 to cardiolipin [140], and are the onset platform for the formation of the NLRP3 immune complex [19]. It has been determined that membrane-associated RING-CH-type finger (MARCH) proteins of E3 ubiquitin ligases are important modulators of immunological responses. The outer membrane protein MARCH5 controls quality control and mitochondrial dynamics [141, 142]. Mice with myeloid cell-specific MARCH5 conditional knockout (cKO) do not release IL-1β and IL-18, resulting in decreased mortality and inflammation during septic shock. Furthermore, the mitochondrial-resident MARCH5 E3 ligase is a possible regulator that initiates NEK7 binding to NLRP3 [19]. Taken together, these findings suggest that mitochondrial damage and dysfunction underlie abnormal follicular development in PCOS by driving the formation of NLRP3 inflammasomes and the subsequent induction of inflammatory signaling pathways (Figure 3).
The mechanism of inflammasome activation regulated by mitochondria in PCOS. PCOS is known for its chronic inflammatory status, and inflammasome activation is an important mechanism, among which the NLRP3 inflammasome has been the most studied. NLRP3 inflammasome activation is regulated by mitochondria in PCOS via mitochondrial ROS regulated by Ca2+ signaling, the TCA cycle and ATP production, glucose and lipid metabolism pathways, mtDNA synthesis regulated by CMPK2, and mitochondrial quality control regulated by MARCH5 and NEK7. This figure was created by Biorender (https://biorender.com/).
5. ER stress and mitochondrial dysfunction in PCOS
5.1 ER stress and PCOS
The ER is the principal organelle responsible for protein biosynthesis, folding, secretion, lipid metabolism, and Ca2+ storage in eukaryotic cells [143, 144]. Under endogenous or exogenous stimuli, ER homeostasis is disrupted, and persistently misfolded or unfolded proteins accumulate in the ER lumen, resulting in the induction of ER stress. Numerous physiological and pathological circumstances, including a breakdown of calcium homeostasis, disruption of redox homeostasis, pathogens, and protein variation, can cause ER stress [145, 146]. ER stress and mitochondria interact closely through multiple mechanisms. ER stress sensors can directly trigger mitochondrial damage and inflammasome assembly. The ultrastructure of mitochondria-associated endoplasmic reticulum (MAM) is a core regulatory hub, and its dysfunction can cause calcium homeostasis imbalance, mitochondrial energy metabolism disorders and a vicious cycle of oxidative stress [147].
Cells initiate the unfolded protein response (UPR), an adaptive mechanism, to cope with ER stress. The three main ER sensors involved in initiating and regulating the UPR are inositol-requiring enzyme 1 (IRE1), protein kinase R (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6) [148, 149]. Under ER stress, the three branches of the UPR (IRE1, PERK, and ATF6) activate several signaling cascades that take part in the regulation of protein synthesis, gene expression, and cell fate decisions such as apoptosis. The three signaling pathways not only have independent feedback loops but also cross-regulatory networks [150-152]. However, the UPR plays a dual role in diseases. It can improve protein folding efficiency and promote ER-related protein degradation and autophagy, thereby eliminating misfolded or unfolded proteins to restore cellular homeostasis and promote cell survival under ER stress. When the ER returns to homeostasis, the UPR receives negative feedback and ER stress stops. On the other hand, when ER stress is prolonged or severe, it leads to cell death [153]. ER stress and the UPR have been implicated in various diseases, including diabetes, obesity, neurodegenerative diseases, cancers, inflammatory diseases, and metabolic disorders [154].
ER stress has been implicated in follicular growth and maturation, follicular atresia, hormone production and secretion, and corpus luteum biogenesis [155-157]. Genes and proteins involved in ER stress have been reported in granulosa cells, oocytes, cumulus-oocyte complexes, theca cells, the corpus luteum, and embryos [158]. There is evidence that ER stress is induced in granulosa cells of women with PCOS and in PCOS mouse model, and that it plays a role in the interplay between inflammation and apoptosis. In human granulosa cells, thapsigargin and tunicamycin, which cause ER stress, raise the production of pro-fibrotic growth factors [159]. A recent study reported increased inflammatory levels, pyroptosis, and ER stress sensor proteins in androgen-induced KGN cells. To investigate the function of ER stress in androgen-induced inflammation and pyroptosis, Xiang et al. treated KGN cells with tauroursodeoxycholic acid (an ER stress inhibitor) and found that suppressing ER stress reduced HA-induced inflammation and pyroptosis. Bioinformatics analyses have identified genes associated with ER stress in PCOS [160]. These results suggest that ER stress may be critical in the imbalanced inflammatory microenvironment of PCOS [161].
5.2 Targeted ER stress therapy in PCOS
Recently, some studies have reported the potential value of inhibiting excessive ER stress in the treatment of PCOS. A randomized clinical trial found that ER stress exists in patients with PCOS and can be alleviated by the administration of astaxanthin (ASX). In this study, patients were classified into ASX or placebo groups, and the results demonstrated that ER stress in granulosa cells of PCOS women could be modulated by ASX. Additionally, the treatment group exhibited higher rates of high-quality oocytes, high-quality embryos, and oocyte maturation, whereas no significant differences were observed in oocyte number, fertilization rate, or fertility rate [162]. Another randomized clinical trial by Jabarpour et al. also found that the ER stress-associated protein ATF6 was upregulated in women with PCOS, and the administration of ASX may benefit PCOS by modulating the ER stress-apoptotic pathway and reducing serum inflammatory marker levels [163]. A further clinical study showed that resveratrol could improve symptoms of patients with PCOS by decreasing pro-inflammatory and ER stress markers [164]. El-Saka et al. found that a PCOS group exhibited increased ER stress after administration of adrenomedullin (ADM) in a rat PCOS model. This can alleviate the disturbances of suppressing of PI3K/Akt1 and PPAR-γ pathways, the imbalance of the sex hormone profile, hyperglycemia, dyslipidemia, IR, increased profibrotic factors, and abnormal ovarian histopathological changes in the ovaries of the PCOS model by attenuating ER stress [165]. Bai et al. showed that IL-18 binding protein (IL-18BP), a specific inhibitory receptor of IL-18, could reverse inflammation, fibrosis, and ER stress-related pathways in PCOS mouse ovaries, and that the level of mitochondrial ROS was also decreased [166]. Electroacupuncture (EA) has also been reported that can alleviated PCOS by regulating ER stress in vitro and in vivo [167, 168]. Furthermore, Peng et al. found that treatment with EA could improve mitochondrial dysfunction by facilitating the activity of complexes I and III in PCOS rats. The above results suggest that targeting and inhibiting ER stress holds a promising therapeutic prospect in PCOS.
5.3 ER stress regulated by mitochondria in PCOS
The pathogenesis of ER stress in PCOS is primarily associated with the UPR triggered by the IRE1, PERK, and ATF6 signaling pathways. Moreover, the ER and mitochondria are closely interconnected and can influence each other. Mitochondria also exhibit UPR after stress. A clinical study found that the UPR genes of the ER, including IREI, ATF6, XBP1, BIP, and CHOP, and the UPR genes of the mitochondria, including HSP10, HSP60, CLPP, and HSP40, were increased in granulosa cells of women with PCOS [169].
ER stress is closely associated with increased mitochondrial ROS levels. Clinical, in vivo, and in vitro studies have reported that PCOS causes ER stress and increases ROS level enhancement [170-172]. The ER and mitochondria interact to ensure cell functional coordination through multiple contact sites, called the MAM, which are involved in the regulation of the intracellular microenvironment, especially in the exchange of ROS and Ca2+. The MAM is tightly connected to the outer membrane of the mitochondria, allowing Ca2+ transfer between the two organelles. ER stress often leads to Ca2+ flow from the ER to the mitochondria via MAMs, resulting in elevated mitochondrial ROS levels, which in turn intensify ER stress and Ca2+ release. This vicious cycle between ER stress and mitochondrial disorder promotes apoptosis [20, 147, 173, 174]. Liao et al. reported that the mitochondrial fusion protein MFN2 regulates MAMs to affect PCOS oocyte development in a mouse model. The expression levels of proteins and genes of mitochondria-related proteins, IP3R, were downregulated in the MAMs of the PCOS model. In order for Ca2+ to be released from the ER into the cytoplasm, IP3R is a crucial Ca2+ release channel in the region. Together with glucose-regulated protein 75 (GRP75) and voltage-dependent anion channel 1 (VDAC1), it forms Ca2+ channel proteins that control Ca2+ transport between the mitochondria and the ER [21]. Yuan et al. used a PCOS cell model to show that intracellular Ca2+ levels increase and induce ER stress [175]. Malfunctioning MAM fail to effectively coordinate mitochondrial biogenesis, dynamics, and quality control, leading to a series of functional disorders, including reduced mitochondrial quantity, abnormal structure, insufficient ATP production, excessive ROS generation and decreased membrane potential. These defects ultimately impair the normal maturation and developmental potential of oocytes and promote the apoptosis of granulosa cells, collectively contributing to abnormal ovarian function and reduced fertility in PCOS [176].
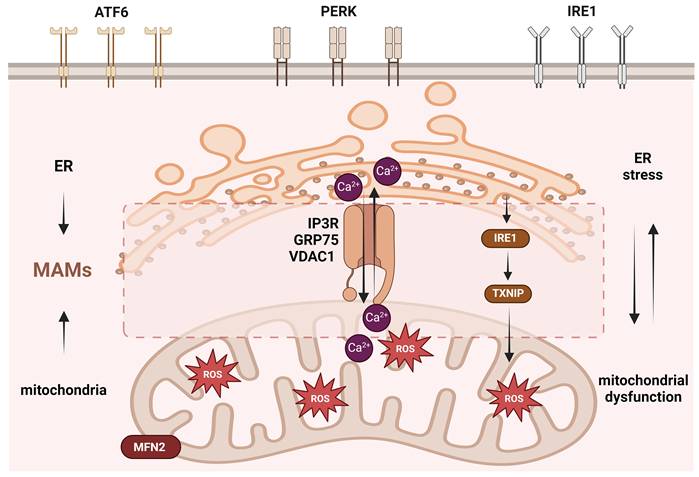
PCOS is a complex process, and ER stress can also regulate ferroptosis and inflammasome activation. Ge et al. demonstrated that the granulosa cells from women with PCOS and from a mouse model exhibited ER stress and mediated ferroptosis in granulosa cells, leading to follicular dysfunction induced by HA. Inhibition of ER stress suppressed HA-induced ferroptosis [177]. Weng et al. found that ROS production in the ovarian cells of a PCOS model induced ER stress and activated the IRE1-TXNIP/ROS-NLRP3 signaling pathway, increasing inflammatory levels [120]. The regulation of MAMs may represent a key mechanism in these processes. MAMs control both Ca²⁺ transfer and ROS signaling, and dysregulation of Ca²⁺ and ROS is known to be closely associated with lipid peroxidation in ferroptosis and the assembly of the NLRP3 inflammasome. As a signaling hub at the interface of these pathways, MAMs likely serve as an important platform that integrates cellular stress, metabolism, and death signals. In ferroptosis, MAM-mediated Ca²⁺ release may exacerbate ER stress and promote the accumulation of lipid peroxidation products, while MAM-derived ROS can directly oxidize polyunsaturated fatty acids, thereby initiating or amplifying the ferroptosis cascade. Regarding the NLRP3 inflammasome, MAM integrity may influence mitochondrial ROS release and Ca²⁺-dependent inflammatory signaling, thereby modulating NLRP3 oligomerization and caspase-1 activation [178, 179]. However, current studies on ER stress caused by mitochondrial dysfunction in PCOS are limited and lack of detailed mechanistic insight (Figure 4).
The mechanism of endoplasmic reticulum stress regulated by mitochondria in PCOS. ER stress is primarily induced by UPR regulation via the IRE1, PERK, and ATF6 pathways. ER stress in PCOS can be regulated by the mitochondria via mitochondrial ROS regulated by IL-18BP; Complex I and Complex III; and by mitochondrial UPR regulated by HSP60, HSP10, CLPP, and HSP40. In addition, the ER and mitochondria have a tightly connected structure called MAMs, which can regulate the exchange of ROS and Ca2+ and can be regulated by the mitochondrial fusion protein MFN2 and Ca2+ release channel protein IP3R. This figure was created by Biorender (https://biorender.com/).
6. Conclusion and future perspectives
PCOS is a systemic, multifactorial, polygenic, metabolic, and inflammatory disease. In this review, we highlight the role of mitochondrial dysfunction in the pathogenesis of PCOS. We propose that three major pathological processes including ferroptosis, inflammasomes, and ER stress, may lead to ovarian dysfunction, metabolic abnormalities, and chronic inflammation in PCOS.
Approximately 10% of women of reproductive age worldwide have PCOS, but 70% of patients fail to be diagnosed early [180]. Research advances have been made in the mechanism of mitochondrial damage, ferroptosis, inflammasome activation, and ER stress in PCOS. However, there are relatively limited reports on predictive indicators of PCOS, especially in the adolescent population. Mitochondria are sensitive organelles that often become abnormal in the early stages of disease, and early markers warrant further exploration and validation. Qasemi et al. reported the potency of cell-free mtDNA levels in follicular fluid as a biomarker for PCOS in women [181]. Another clinical study reported the predictive value of the oxidative stress-associated biomarker 8-Isoprostane [182]. However, the sample size of this study was small.
Currently, there is a lack of early predictive indicators and reference ranges that can be widely applied in clinical practice. For high-risk women of reproductive age who are obese or have diabetes, early detection and intervention based on appropriate predictive indicators could significantly reduce the incidence of PCOS and its adverse outcomes. This represents an important direction for future research on PCOS.
Abbreviations
ACSL4: synthetase long-chain family member 4; ADM: adrenomedullin; AhR: aryl hydrocarbon receptor; ATF6: activating transcription factor 6; ATP: adenosine triphosphate; ASC: apoptosis-associated speck-like protein; ASX: astaxanthin; BNIP3L: the BCL2/adenovirus E1B interacting protein 3-like; CISD2: CDGSH iron sulfur domain 2; CLPP: caseinolytic protease P; CMPK2: cytidine/uridine monophosphate kinase 2; CoA: acyl-coenzyme A; DEHP: di-(2-ethylhexyl) phthalate; DHEA: dehydroepiandrosterone; DHT: dihydrotestosterone; Dpp4: dipeptidyl peptidase-4; DPP-IV: dipeptidyl peptidase-IV; DRP1: dynamin-related protein 1; EA: electroacupuncture; ER: endoplasmic reticulum; ERK: extracellular signal-regulated kinase; ETC: electron transport chain; FAC: ferric ammonium citrate; Fer-1: ferrostatin-1; Fis1: mitochondrial fission 1 protein; Foxo1: Forkhead Box Protein O1; FTH1: ferritin heavy chain 1; FUNDC1: FUN14 domain containing 1; GLUT1: glucose trans porter-1; GPX4: glutathione peroxidase 4; GRP75: Glucose-regulated protein 75; GSDMD: activated caspase-1 cleaves gasdermin D; GSH: glutathione; HA: Hyperandrogenism; IR: insulin resistance; HAIR: hepatocellular adenoma with iron-related disease; HDAC1: Histone deacetylase 1; HIF-1α: hypoxia-inducible factor-1 alpha; HK1: hexokinase 1; H2O2: hydrogen peroxide; HSP60: heat shock protein 60; IL-18BP: IL-18 binding protein; IP3: inositol 1,4,5-triphosphate receptor; IPA: indole-3-propionic acid; IRE1: inositol-requiring enzyme 1; JNK: c-Jun N-terminal kinase; LDHA: lactate dehydrogenase A; mt-tRNA: mitochondrial transfer RNA; MAMs: mitochondria-associated endoplasmic reticulum; MAPK: mitogen-activated protein kinase; MARCH5: mitochondrial ubiquitin ligase membrane-associated RING-CH; MFF: mitochondrial fission factor; MFN2: mitofusin-2; mtDNA: mitochondrial DNA; MQC: mitochondrial quality control; NCOA4: nuclear receptor coactivator 4; NEK7: NIMA-related kinase 7; NLRP3: NLR family pyrin domain containing 3; NOX1: NADPH oxidase 1; Nrf2: NF-E2-related factor 2; OPA1: optic atrophy 1; PCOS: polycystic ovary syndrome; PERK: protein kinase R (PKR)-like ER kinase; PFK: phosphofructokinase; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator; PINK: PTEN-induced kinase; PLOHs: phospholipid alcohols; PLOOHs: phospholipid hydroperoxides; RBP4: retinol-binding protein 4; ROS: reactive oxygen species; SIRT1: Sirtuin 1; SIRT3: Sirtuin 3; SOD1: superoxide dismutase 1; TCA: tricarboxylic acid; TFAM: mitochondrial transcription factor A; UPR: unfolded protein response; VDAC1: voltage-dependent anion channel 1; WTAP: Wilms' tumor 1-associated protein.
Acknowledgements
I am really appreciative of and would like to express my gratitude to Professor Jichun Tan and Liu Cao for the advice and assistance in completing this project.
Funding
The National Natural Science Foundation of China (No. 82201810), the Shengjing Freelance Researcher Plan of Shengjing Hospital of China Medical University and the National Key Research and Development Program (2024YFC2706702).
Authors contributions
Jichun Tan and Liu Cao revised the manuscript; Xinyi Zhang wrote the original manuscript; Tong sun drew the figures; Xinxin Wang and Yujiu Ma searched the literature.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Teede H, Deeks A, Moran L. Polycystic ovary syndrome: a complex condition with psychological, reproductive and metabolic manifestations that impacts on health across the lifespan. BMC medicine. 2010;8:41
2. Teede HJ, Tay CT, Laven JJE, Dokras A, Moran LJ, Piltonen TT. et al. Recommendations from the 2023 international evidence-based guideline for the assessment and management of polycystic ovary syndrome. European journal of endocrinology. 2023;189:G43-g64
3. Goodarzi MO, Dumesic DA, Chazenbalk G, Azziz R. Polycystic ovary syndrome: etiology, pathogenesis and diagnosis. Nature reviews Endocrinology. 2011;7:219-31
4. Hillman SC, Dale J. Polycystic ovarian syndrome: an under-recognised problem? The British journal of general practice: the journal of the Royal College of General Practitioners. 2018;68:244
5. Fang Y, Liu L, Yang Y, Zhang B, Xie S. Causal Association Between BMI and Polycystic Ovarian Syndrome: Bidirectional 2-Sample Mendelian Randomization Study. The Journal of clinical endocrinology and metabolism. 2024;110:41-7
6. Carmina E. Reproductive System Outcome Among Patients with Polycystic Ovarian Syndrome. Endocrinology and metabolism clinics of North America. 2015;44:787-97
7. Qiu M, Qu J, Tian Y, Wang Y. The influence of polycystic ovarian syndrome on obstetric and neonatal outcomes after frozen-thawed embryo transfer. Reproductive biomedicine online. 2022;45:745-53
8. Bahri Khomami M, Shorakae S, Hashemi S, Harrison CL, Piltonen TT, Romualdi D. et al. Systematic review and meta-analysis of pregnancy outcomes in women with polycystic ovary syndrome. Nature communications. 2024;15:5591
9. Millán-de-Meer M, Luque-Ramírez M, Nattero-Chávez L, Escobar-Morreale HF. PCOS during the menopausal transition and after menopause: a systematic review and meta-analysis. Human reproduction update. 2023;29:741-72
10. Pace L, Markovic D, Buyalos R, Bril F, Azziz R. Economic Burden of Endometrial Cancer Associated With Polycystic Ovary Syndrome. The Journal of clinical endocrinology and metabolism. 2024;110:e168-e76
11. Dason ES, Koshkina O, Chan C, Sobel M. Diagnosis and management of polycystic ovarian syndrome. CMAJ: Canadian Medical Association journal = journal de l'Association medicale canadienne. 2024;196:E85-e94
12. Senthilkumar H, Chauhan SC, Arumugam M. Unraveling the multifactorial pathophysiology of polycystic ovary syndrome: exploring lifestyle, prenatal influences, neuroendocrine dysfunction, and post-translational modifications. Molecular biology reports. 2025;52:980
13. Singh I, Moar K, Maurya PK. Diagnostic and prognostic biomarkers in Polycystic Ovary Syndrome. Clinica chimica acta; international journal of clinical chemistry. 2025;576:120425
14. Gibson-Helm M, Teede H, Dunaif A, Dokras A. Delayed Diagnosis and a Lack of Information Associated With Dissatisfaction in Women With Polycystic Ovary Syndrome. The Journal of clinical endocrinology and metabolism. 2017;102:604-12
15. Hoeger KM, Dokras A, Piltonen T. Update on PCOS: Consequences, Challenges, and Guiding Treatment. The Journal of clinical endocrinology and metabolism. 2021;106:e1071-e83
16. Kotlyar AM, Seifer DB. Women with PCOS who undergo IVF: a comprehensive review of therapeutic strategies for successful outcomes. Reproductive biology and endocrinology: RB&E. 2023;21:70
17. Dabravolski SA, Nikiforov NG, Eid AH, Nedosugova LV, Starodubova AV, Popkova TV. et al. Mitochondrial Dysfunction and Chronic Inflammation in Polycystic Ovary Syndrome. International journal of molecular sciences. 2021 22
18. Siemers KM, Klein AK, Baack ML. Mitochondrial Dysfunction in PCOS: Insights into Reproductive Organ Pathophysiology. International journal of molecular sciences. 2023 24
19. Park YJ, Dodantenna N, Kim Y, Kim TH, Lee HS, Yoo YS. et al. MARCH5-dependent NLRP3 ubiquitination is required for mitochondrial NLRP3-NEK7 complex formation and NLRP3 inflammasome activation. The EMBO journal. 2023;42:e113481
20. Liu YT, Zhang H, Duan SB, Wang JW, Chen H, Zhan M. et al. Mitofusin2 Ameliorated Endoplasmic Reticulum Stress and Mitochondrial Reactive Oxygen Species Through Maintaining Mitochondria-Associated Endoplasmic Reticulum Membrane Integrity in Cisplatin-Induced Acute Kidney Injury. Antioxidants & redox signaling. 2024;40:16-39
21. Liao X, Zhu S, Qiu S, Cao H, Jiang W, Xu H. et al. Mfn2 regulates mitochondria-associated ER membranes to affect PCOS oocyte development. Endocrine connections. 2024 13
22. Palma FR, Gantner BN, Sakiyama MJ, Kayzuka C, Shukla S, Lacchini R. et al. ROS production by mitochondria: function or dysfunction? Oncogene. 2024;43:295-303
23. Madreiter-Sokolowski CT, Thomas C, Ristow M. Interrelation between ROS and Ca(2+) in aging and age-related diseases. Redox biology. 2020;36:101678
24. Zhang B, Pan C, Feng C, Yan C, Yu Y, Chen Z. et al. Role of mitochondrial reactive oxygen species in homeostasis regulation. Redox report: communications in free radical research. 2022;27:45-52
25. Andrieux P, Chevillard C, Cunha-Neto E, Nunes JPS. Mitochondria as a Cellular Hub in Infection and Inflammation. International journal of molecular sciences. 2021 22
26. Zhang R, Liu H, Bai H, Zhang Y, Liu Q, Guan L. et al. Oxidative stress status in Chinese women with different clinical phenotypes of polycystic ovary syndrome. Clinical endocrinology. 2017;86:88-96
27. Wang YC, Ma YD, Liu H, Cui ZH, Zhao D, Zhang XQ. et al. Hyperandrogen-induced polyol pathway flux increase affects ovarian function in polycystic ovary syndrome via excessive oxidative stress. Life sciences. 2023;313:121224
28. Konopka AR, Asante A, Lanza IR, Robinson MM, Johnson ML, Dalla Man C. et al. Defects in mitochondrial efficiency and H2O2 emissions in obese women are restored to a lean phenotype with aerobic exercise training. Diabetes. 2015;64:2104-15
29. Victor VM, Rocha M, Bañuls C, Alvarez A, de Pablo C, Sanchez-Serrano M. et al. Induction of oxidative stress and human leukocyte/endothelial cell interactions in polycystic ovary syndrome patients with insulin resistance. The Journal of clinical endocrinology and metabolism. 2011;96:3115-22
30. Victor VM, Rovira-Llopis S, Bañuls C, Diaz-Morales N, Castelló R, Falcón R. et al. Effects of metformin on mitochondrial function of leukocytes from polycystic ovary syndrome patients with insulin resistance. European journal of endocrinology. 2015;173:683-91
31. Hu M, Zhang Y, Guo X, Jia W, Liu G, Zhang J. et al. Hyperandrogenism and insulin resistance induce gravid uterine defects in association with mitochondrial dysfunction and aberrant reactive oxygen species production. American journal of physiology Endocrinology and metabolism. 2019;316:E794-e809
32. Zhang Y, Zhao W, Xu H, Hu M, Guo X, Jia W. et al. Hyperandrogenism and insulin resistance-induced fetal loss: evidence for placental mitochondrial abnormalities and elevated reactive oxygen species production in pregnant rats that mimic the clinical features of polycystic ovary syndrome. The Journal of physiology. 2019;597:3927-50
33. Luo M, Zheng LW, Wang YS, Huang JC, Yang ZQ, Yue ZP. et al. Genistein exhibits therapeutic potential for PCOS mice via the ER-Nrf2-Foxo1-ROS pathway. Food & function. 2021;12:8800-11
34. Kobayashi M, Yoshino O, Nakashima A, Ito M, Nishio K, Ono Y. et al. Inhibition of autophagy in theca cells induces CYP17A1 and PAI-1 expression via ROS/p38 and JNK signalling during the development of polycystic ovary syndrome. Molecular and cellular endocrinology. 2020;508:110792
35. Costa J, Braga PC, Rebelo I, Oliveira PF, Alves MG. Mitochondria Quality Control and Male Fertility. Biology. 2023 12
36. Boyman L, Karbowski M, Lederer WJ. Regulation of Mitochondrial ATP Production: Ca(2+) Signaling and Quality Control. Trends in molecular medicine. 2020;26:21-39
37. Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nature cell biology. 2018;20:745-54
38. Zhang CH, Liu XY, Wang J. Essential Role of Granulosa Cell Glucose and Lipid Metabolism on Oocytes and the Potential Metabolic Imbalance in Polycystic Ovary Syndrome. International journal of molecular sciences. 2023 24
39. Pokorska-Niewiada K, Brodowska A, Szczuko M. The Content of Minerals in the PCOS Group and the Correlation with the Parameters of Metabolism. Nutrients. 2021 13
40. Zhao YK, Gao YN, Wang LC, Wang J, Wang GJ, Wu HL. Correlation between abnormal energy metabolism of ovarian granulosa cells and in vitro fertilization-embryo transfer outcomes in patients with polycystic ovary syndrome and obesity. Journal of ovarian research. 2023;16:145
41. Mazloomi S, Farimani MS, Tavilani H, Karimi J, Amiri I, Abbasi E. et al. Granulosa cells from immature follicles exhibit restricted glycolysis and reduced energy production: a dominant problem in polycystic ovary syndrome. Journal of assisted reproduction and genetics. 2023;40:343-59
42. Zhang Q, Ren J, Wang F, Pan M, Cui L, Li M. et al. Mitochondrial and glucose metabolic dysfunctions in granulosa cells induce impaired oocytes of polycystic ovary syndrome through Sirtuin 3. Free radical biology & medicine. 2022;187:1-16
43. Cao J, Huo P, Cui K, Wei H, Cao J, Wang J. et al. Follicular fluid-derived exosomal miR-143-3p/miR-155-5p regulate follicular dysplasia by modulating glycolysis in granulosa cells in polycystic ovary syndrome. Cell communication and signaling: CCS. 2022;20:61
44. Ma H, Qi D, Xu Y, Shang T, Si Y, Chen W. et al. PLK2 as a key regulator of glycolysis and immune dysregulation in polycystic ovary syndrome. Frontiers in immunology. 2025;16:1610713
45. Pfanner N, Warscheid B, Wiedemann N. Mitochondrial proteins: from biogenesis to functional networks. Nat Rev Mol Cell Biol. 2019;20:267-84
46. Chen M, Yan R, Luo J, Ning J, Zhou R, Ding L. The Role of PGC-1α-Mediated Mitochondrial Biogenesis in Neurons. Neurochemical research. 2023;48:2595-606
47. Sharma P, Sampath H. Mitochondrial DNA Integrity: Role in Health and Disease. Cells. 2019 8
48. Ardehjani NA, Agha-Hosseini M, Nashtaei MS, Khodarahmian M, Shabani M, Jabarpour M. et al. Resveratrol ameliorates mitochondrial biogenesis and reproductive outcomes in women with polycystic ovary syndrome undergoing assisted reproduction: a randomized, triple-blind, placebo-controlled clinical trial. Journal of ovarian research. 2024;17:143
49. Safaei Z, Bakhshalizadeh S, Nasr-Esfahani MH, Akbari Sene A, Najafzadeh V, Soleimani M. et al. Vitamin D3 affects mitochondrial biogenesis through mitogen-activated protein kinase in polycystic ovary syndrome mouse model. Journal of cellular physiology. 2020;235:6113-26
50. Sun L, Tian H, Xue S, Ye H, Xue X, Wang R. et al. Circadian Clock Genes REV-ERBs Inhibits Granulosa Cells Apoptosis by Regulating Mitochondrial Biogenesis and Autophagy in Polycystic Ovary Syndrome. Frontiers in cell and developmental biology. 2021;9:658112
51. Pang X, Cheng J, Wu T, Sun L. SIRT3 ameliorates polycystic ovary syndrome through FOXO1/PGC-1α signaling pathway. Endocrine. 2023;80:201-11
52. Abu Shelbayeh O, Arroum T, Morris S, Busch KB. PGC-1α Is a Master Regulator of Mitochondrial Lifecycle and ROS Stress Response. Antioxidants (Basel, Switzerland). 2023 12
53. Lin DS, Huang YW, Ho CS, Hung PL, Hsu MH, Wang TJ. et al. Oxidative Insults and Mitochondrial DNA Mutation Promote Enhanced Autophagy and Mitophagy Compromising Cell Viability in Pluripotent Cell Model of Mitochondrial Disease. Cells. 2019 8
54. Tharayil SP, Rasal S, Gawde U, Mukherjee S, Patil A, Joshi B. et al. Relation of mitochondrial DNA copy number and variants with the clinical characteristics of polycystic ovary syndrome. Molecular and cellular endocrinology. 2024;594:112386
55. Ding Y, Zhuo G, Zhang C, Leng J. Point mutation in mitochondrial tRNA gene is associated with polycystic ovary syndrome and insulin resistance. Molecular medicine reports. 2016;13:3169-72
56. Ding Y, Xia BH, Zhang CJ, Zhuo GC. Mitochondrial tRNA(Leu(UUR)) C3275T, tRNA(Gln) T4363C and tRNA(Lys) A8343G mutations may be associated with PCOS and metabolic syndrome. Gene. 2018;642:299-306
57. Atherton W, Ambrose L, Wisdom J, Lessard BD, Kurucz N, Webb CE. et al. Nuclear and mitochondrial population genetics of the Australasian arbovirus vector Culex annulirostris (Skuse) reveals strong geographic structure and cryptic species. Parasites & vectors. 2024;17:501
58. Chen Y, Wu WJ, Xing LW, Zhang XJ, Wang J, Xia XY. et al. Investigating the role of mitochondrial DNA D-loop variants, haplotypes, and copy number in polycystic ovary syndrome: implications for clinical phenotypes in the Chinese population. Frontiers in endocrinology. 2023;14:1206995
59. Nawaz T, Awan T, Zahoor H, Gul R, Bibi S, Uddin A. et al. Analysis of mutations in mitochondrial transfer RNA genes and the maternal inheritance of polycystic ovary syndrome. Frontiers in endocrinology. 2025;16:1509791
60. Chan DC. Mitochondrial Dynamics and Its Involvement in Disease. Annual review of pathology. 2020;15:235-59
61. Chandhok G, Lazarou M, Neumann B. Structure, function, and regulation of mitofusin-2 in health and disease. Biological reviews of the Cambridge Philosophical Society. 2018;93:933-49
62. Adebayo M, Singh S, Singh AP, Dasgupta S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2021;35:e21620
63. Salehi R, Mazier HL, Nivet AL, Reunov AA, Lima P, Wang Q. et al. Ovarian mitochondrial dynamics and cell fate regulation in an androgen-induced rat model of polycystic ovarian syndrome. Scientific reports. 2020;10:1021
64. Li X, He Y, Yan Q, Kuai D, Zhang H, Wang Y. et al. Dihydrotestosterone induces reactive oxygen species accumulation and mitochondrial fission leading to apoptosis of granulosa cells. Toxicology. 2024;509:153958
65. Yan MQ, Wang Y, Wang Z, Liu XH, Yang YM, Duan XY. et al. Mitoguardin2 Is Associated With Hyperandrogenism and Regulates Steroidogenesis in Human Ovarian Granulosa Cells. Journal of the Endocrine Society. 2023;7:bvad034
66. Li H, Liu J, Huang L, Cheng Y, Zhao G, Pan X. et al. Hexavalent chromium inhibits testosterone synthesis in bovine testicular leydig cells through mitochondrial damage via BNIP3. Reproductive biology. 2026;26:101181
67. Wang S, Long H, Hou L, Feng B, Ma Z, Wu Y. et al. The mitophagy pathway and its implications in human diseases. Signal transduction and targeted therapy. 2023;8:304
68. Lu Y, Li Z, Zhang S, Zhang T, Liu Y, Zhang L. Cellular mitophagy: Mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics. 2023;13:736-66
69. Wu HH, Zhu Q, Liang N, Xiang Y, Xu TY, Huang ZC. et al. CISD2 regulates oxidative stress and mitophagy to maintain the balance of the follicular microenvironment in PCOS. Redox report: communications in free radical research. 2024;29:2377870
70. Yi S, Zheng B, Zhu Y, Cai Y, Sun H, Zhou J. Melatonin ameliorates excessive PINK1/Parkin-mediated mitophagy by enhancing SIRT1 expression in granulosa cells of PCOS. American journal of physiology Endocrinology and metabolism. 2020;319:E91-e101
71. Wang F, Han J, Wang X, Liu Y, Zhang Z. Roles of HIF-1α/BNIP3 mediated mitophagy in mitochondrial dysfunction of letrozole-induced PCOS rats. Journal of molecular histology. 2022;53:833-42
72. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060-72
73. Stockwell BR. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185:2401-21
74. Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Molecular cell. 2022;82:2215-27
75. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266-82
76. Wang X, Zhou Y, Min J, Wang F. Zooming in and out of ferroptosis in human disease. Front Med. 2023;17:173-206
77. Qin X, Liang D, Hu M, Lv Z, Duan Z, Liu X. et al. Chronic overload of concentration-dependent iron exerts different effects on ovarian function in C57BL/6J mice†. Biology of reproduction. 2021;104:1347-59
78. Sze SCW, Zhang L, Zhang S, Lin K, Ng TB, Ng ML. et al. Aberrant Transferrin and Ferritin Upregulation Elicits Iron Accumulation and Oxidative Inflammaging Causing Ferroptosis and Undermines Estradiol Biosynthesis in Aging Rat Ovaries by Upregulating NF-Κb-Activated Inducible Nitric Oxide Synthase: First Demonstration of an Intricate Mechanism. International journal of molecular sciences. 2022 23
79. Zhou J, Lin L, Liu L, Wang J, Xia G, Wang C. The transcriptome reveals the molecular regulatory network of primordial follicle depletion in obese mice. Fertility and sterility. 2023;120:899-910
80. Hu J, Wang H, Fang J, Jiang R, Kong Y, Zhang T. et al. Ovarian aging-associated downregulation of GPX4 expression regulates ovarian follicular development by affecting granulosa cell functions and oocyte quality. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2025;39:e70469
81. Kim JW, Kang KM, Yoon TK, Shim SH, Lee WS. Study of circulating hepcidin in association with iron excess, metabolic syndrome, and BMP-6 expression in granulosa cells in women with polycystic ovary syndrome. Fertility and sterility. 2014;102:548-54.e2
82. Martínez-García MA, Luque-Ramírez M, San-Millán JL, Escobar-Morreale HF. Body iron stores and glucose intolerance in premenopausal women: role of hyperandrogenism, insulin resistance, and genomic variants related to inflammation, oxidative stress, and iron metabolism. Diabetes care. 2009;32:1525-30
83. Zhang Y, Hu M, Jia W, Liu G, Zhang J, Wang B. et al. Hyperandrogenism and insulin resistance modulate gravid uterine and placental ferroptosis in PCOS-like rats. The Journal of endocrinology. 2020;246:247-63
84. Hu M, Zhang Y, Ma S, Li J, Wang X, Liang M. et al. Suppression of uterine and placental ferroptosis by N-acetylcysteine in a rat model of polycystic ovary syndrome. Molecular human reproduction. 2021 27
85. Zhang D, Yi S, Cai B, Wang Z, Chen M, Zheng Z. et al. Involvement of ferroptosis in the granulosa cells proliferation of PCOS through the circRHBG/miR-515/SLC7A11 axis. Annals of translational medicine. 2021;9:1348
86. Tan W, Dai F, Yang D, Deng Z, Gu R, Zhao X. et al. MiR-93-5p promotes granulosa cell apoptosis and ferroptosis by the NF-kB signaling pathway in polycystic ovary syndrome. Frontiers in immunology. 2022;13:967151
87. Li X, Lin Y, Cheng X, Yao G, Yao J, Hu S. et al. Ovarian ferroptosis induced by androgen is involved in pathogenesis of PCOS. Human reproduction open. 2024;2024:hoae013
88. Huang J, Fan H, Li C, Yang K, Xiong C, Xiong S. et al. Dysregulation of ferroptosis-related genes in granulosa cells associates with impaired oocyte quality in polycystic ovary syndrome. Frontiers in endocrinology. 2024;15:1346842
89. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB. et al. Role of Mitochondria in Ferroptosis. Molecular cell. 2019;73:354-63.e3
90. Wang H, Liu C, Zhao Y, Gao G. Mitochondria regulation in ferroptosis. European journal of cell biology. 2020;99:151058
91. Gan B. Mitochondrial regulation of ferroptosis. The Journal of cell biology. 2021 220
92. Zheng J, Conrad M. The Metabolic Underpinnings of Ferroptosis. Cell metabolism. 2020;32:920-37
93. Zhang L, Wang F, Li D, Yan Y, Wang H. Transferrin receptor-mediated reactive oxygen species promotes ferroptosis of KGN cells via regulating NADPH oxidase 1/PTEN induced kinase 1/acyl-CoA synthetase long chain family member 4 signaling. Bioengineered. 2021;12:4983-94
94. Li YY, Peng YQ, Yang YX, Shi TJ, Liu RX, Luan YY. et al. Baicalein improves the symptoms of polycystic ovary syndrome by mitigating oxidative stress and ferroptosis in the ovary and gravid placenta. Phytomedicine: international journal of phytotherapy and phytopharmacology. 2024;128:155423
95. Zhang P, Pan Y, Wu S, He Y, Wang J, Chen L. et al. n-3 PUFA Promotes Ferroptosis in PCOS GCs by Inhibiting YAP1 through Activation of the Hippo Pathway. Nutrients. 2023 15
96. Li C, Dong X, Du W, Shi X, Chen K, Zhang W. et al. LKB1-AMPK axis negatively regulates ferroptosis by inhibiting fatty acid synthesis. Signal transduction and targeted therapy. 2020;5:187
97. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Molecular cell. 2015;59:298-308
98. Kerins MJ, Ooi A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxidants & redox signaling. 2018;29:1756-73
99. Skonieczna M, Cieslar-Pobuda A, Saenko Y, Foksinski M, Olinski R, Rzeszowska-Wolny J. et al. The Impact of DIDS-Induced Inhibition of Voltage-Dependent Anion Channels (VDAC) on Cellular Response of Lymphoblastoid Cells to Ionizing Radiation. Medicinal chemistry (Shariqah (United Arab Emirates)). 2017;13:477-83
100. Li J, Jia YC, Ding YX, Bai J, Cao F, Li F. The crosstalk between ferroptosis and mitochondrial dynamic regulatory networks. International journal of biological sciences. 2023;19:2756-71
101. Catapano A, Cimmino F, Petrella L, Pizzella A, D'Angelo M, Ambrosio K. et al. Iron metabolism and ferroptosis in health and diseases: The crucial role of mitochondria in metabolically active tissues. The Journal of nutritional biochemistry. 2025;140:109888
102. Patel S. Polycystic ovary syndrome (PCOS), an inflammatory, systemic, lifestyle endocrinopathy. The Journal of steroid biochemistry and molecular biology. 2018;182:27-36
103. Popovic M, Sartorius G, Christ-Crain M. Chronic low-grade inflammation in polycystic ovary syndrome: is there a (patho)-physiological role for interleukin-1? Seminars in immunopathology. 2019;41:447-59
104. Rudnicka E, Suchta K, Grymowicz M, Calik-Ksepka A, Smolarczyk K, Duszewska AM. et al. Chronic Low Grade Inflammation in Pathogenesis of PCOS. International journal of molecular sciences. 2021 22
105. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nature medicine. 2015;21:677-87
106. Rostamtabar M, Esmaeilzadeh S, Tourani M, Rahmani A, Baee M, Shirafkan F. et al. Pathophysiological roles of chronic low-grade inflammation mediators in polycystic ovary syndrome. Journal of cellular physiology. 2021;236:824-38
107. Liu Y, Liu H, Li Z, Fan H, Yan X, Liu X. et al. The Release of Peripheral Immune Inflammatory Cytokines Promote an Inflammatory Cascade in PCOS Patients via Altering the Follicular Microenvironment. Frontiers in immunology. 2021;12:685724
108. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular cell. 2002;10:417-26
109. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013-22
110. Barnett KC, Li S, Liang K, Ting JP. A 360° view of the inflammasome: Mechanisms of activation, cell death, and diseases. Cell. 2023;186:2288-312
111. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660-5
112. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. International journal of molecular sciences. 2019 20
113. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nature reviews Immunology. 2019;19:477-89
114. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666-71
115. Chen X, He WT, Hu L, Li J, Fang Y, Wang X. et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell research. 2016;26:1007-20
116. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nature reviews Immunology. 2016;16:407-20
117. Maier NK, Crown D, Liu J, Leppla SH, Moayeri M. Arsenic trioxide and other arsenical compounds inhibit the NLRP1, NLRP3, and NAIP5/NLRC4 inflammasomes. Journal of immunology (Baltimore, Md: 1950). 2014;192:763-70
118. He Y, Hara H, Núñez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends in biochemical sciences. 2016;41:1012-21
119. Wang B, Shi M, Yu C, Pan H, Shen H, Du Y. et al. NLRP3 Inflammasome-dependent Pathway is Involved in the Pathogenesis of Polycystic Ovary Syndrome. Reproductive sciences (Thousand Oaks, Calif). 2024;31:1017-27
120. Weng Y, Zhang Y, Wang D, Wang R, Xiang Z, Shen S. et al. Exercise-induced irisin improves follicular dysfunction by inhibiting IRE1α-TXNIP/ROS-NLRP3 pathway in PCOS. Journal of ovarian research. 2023;16:151
121. Rostamtabar M, Esmaeilzadeh S, Karkhah A, Amiri M, Rahmani A, Bakouei F. et al. Elevated expression of IL-18 but not IL-1β gene is associated with NALP3 and AIM2 inflammasome in Polycystic Ovary Syndrome. Gene. 2020;731:144352
122. Liu Z, Wang RH, Wang KH. Formononetin ameliorates polycystic ovary syndrome through suppressing NLRP3 inflammasome. Molecular medicine (Cambridge, Mass). 2025;31:27
123. Li Z, Geng H, Ye C, Cao L, Qin R, Chen K. et al. Gut microbial metabolite indole-3-propionic acid alleviates polycystic ovary syndrome in mice by regulating the AhR-NLRP3 axis. International immunopharmacology. 2025;148:114038
124. Alenezi SA, Khan R, Snell L, Aboeldalyl S, Amer S. The Role of NLRP3 Inflammasome in Obesity and PCOS-A Systematic Review and Meta-Analysis. International journal of molecular sciences. 2023 24
125. Lai Y, Ye Z, Mu L, Zhang Y, Long X, Zhang C. et al. Elevated Levels of Follicular Fatty Acids Induce Ovarian Inflammation via ERK1/2 and Inflammasome Activation in PCOS. The Journal of clinical endocrinology and metabolism. 2022;107:2307-17
126. Sun J, Gan L, Lv S, Wang T, Dai C, Sun J. Exposure to Di-(2-Ethylhexyl) phthalate drives ovarian dysfunction by inducing granulosa cell pyroptosis via the SLC39A5/NF-κB/NLRP3 axis. Ecotoxicology and environmental safety. 2023;252:114625
127. Zou J, Zheng Y, Huang Y, Tang D, Kang R, Chen R. The Versatile Gasdermin Family: Their Function and Roles in Diseases. Frontiers in immunology. 2021;12:751533
128. Wang D, Weng Y, Zhang Y, Wang R, Wang T, Zhou J. et al. Exposure to hyperandrogen drives ovarian dysfunction and fibrosis by activating the NLRP3 inflammasome in mice. The Science of the total environment. 2020;745:141049
129. Huang J, Chen P, Xiang Y, Liang Q, Wu T, Liu J. et al. Gut microbiota dysbiosis-derived macrophage pyroptosis causes polycystic ovary syndrome via steroidogenesis disturbance and apoptosis of granulosa cells. International immunopharmacology. 2022;107:108717
130. Cai Z, He S, Liu R, Zhou L, Zhao L. Plumbagin rescues the granulosa cell's pyroptosis by reducing WTAP-mediated N6-methylation in polycystic ovary syndrome. Journal of ovarian research. 2022;15:126
131. Chen J, Zhu Z, Xu S, Li J, Huang L, Tan W. et al. HDAC1 participates in polycystic ovary syndrome through histone modification by regulating H19/miR-29a-3p/NLRP3-mediated granulosa cell pyroptosis. Molecular and cellular endocrinology. 2023;573:111950
132. Wu P, Zhu Y, Li J, Chen H, Wu H, Hu X. et al. Guizhi Fuling Wan inhibits autophagy of granulosa cells in polycystic ovary syndrome mice via H19/miR-29b-3p. Gynecological endocrinology: the official journal of the International Society of Gynecological Endocrinology. 2023;39:2210232
133. Li Y, Yao N, Gao Y, Wang Y, Bai L, Xu J. et al. MiR-1224-5p attenuates polycystic ovary syndrome through inhibiting NOD-like receptor protein 3 inflammasome activation via targeting Forkhead box O 1. Bioengineered. 2021;12:8555-69
134. Elliott EI, Miller AN, Banoth B, Iyer SS, Stotland A, Weiss JP. et al. Cutting Edge: Mitochondrial Assembly of the NLRP3 Inflammasome Complex Is Initiated at Priming. Journal of immunology (Baltimore, Md: 1950). 2018;200:3047-52
135. Minutoli L, Puzzolo D, Rinaldi M, Irrera N, Marini H, Arcoraci V. et al. ROS-Mediated NLRP3 Inflammasome Activation in Brain, Heart, Kidney, and Testis Ischemia/Reperfusion Injury. Oxidative medicine and cellular longevity. 2016;2016:2183026
136. Paik S, Kim JK, Silwal P, Sasakawa C, Jo EK. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cellular & molecular immunology. 2021;18:1141-60
137. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ. et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560:198-203
138. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S. et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401-14
139. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC. et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature immunology. 2011;12:222-30
140. Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK. et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39:311-23
141. Lin H, Li S, Shu HB. The Membrane-Associated MARCH E3 Ligase Family: Emerging Roles in Immune Regulation. Frontiers in immunology. 2019;10:1751
142. Shiiba I, Takeda K, Nagashima S, Yanagi S. Overview of Mitochondrial E3 Ubiquitin Ligase MITOL/MARCH5 from Molecular Mechanisms to Diseases. International journal of molecular sciences. 2020 21
143. Schwarz DS, Blower MD. The endoplasmic reticulum: structure, function and response to cellular signaling. Cellular and molecular life sciences: CMLS. 2016;73:79-94
144. Almanza A, Carlesso A, Chintha C, Creedican S, Doultsinos D, Leuzzi B. et al. Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications. The FEBS journal. 2019;286:241-78
145. Chen X, Cubillos-Ruiz JR. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nature reviews Cancer. 2021;21:71-88
146. Chen X, Shi C, He M, Xiong S, Xia X. Endoplasmic reticulum stress: molecular mechanism and therapeutic targets. Signal transduction and targeted therapy. 2023;8:352
147. Mao H, Chen W, Chen L, Li L. Potential role of mitochondria-associated endoplasmic reticulum membrane proteins in diseases. Biochemical pharmacology. 2022;199:115011
148. Deng B, Liao F, Liu Y, He P, Wei S, Liu C. et al. Comprehensive analysis of endoplasmic reticulum stress-associated genes signature of ulcerative colitis. Frontiers in immunology. 2023;14:1158648
149. Yan T, Ma X, Guo L, Lu R. Targeting endoplasmic reticulum stress signaling in ovarian cancer therapy. Cancer biology & medicine. 2023;20:748-64
150. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M. et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Molecular cell. 2000;6:1099-108
151. Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B. et al. IRE1 signaling affects cell fate during the unfolded protein response. Science (New York, NY). 2007;318:944-9
152. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519-29
153. Huang N, Yu Y, Qiao J. Dual role for the unfolded protein response in the ovary: adaption and apoptosis. Protein & cell. 2017;8:14-24
154. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21:421-38
155. Shaoyong W, Liu Y, Xu B, Pan B, Xianmi X, Wang Y. et al. Exposure to BDE-47 causes female infertility risk and induces oxidative stress and lipotoxicity-mediated ovarian hormone secretion disruption in mice. The Science of the total environment. 2022;842:156885
156. Li H, Jing Y, Qu X, Yang J, Pan P, Liu X. et al. The Activation of Reticulophagy by ER Stress through the ATF4-MAP1LC3A-CCPG1 Pathway in Ovarian Granulosa Cells Is Linked to Apoptosis and Necroptosis. International journal of molecular sciences. 2023 24
157. Zhu H, Li X, Qiao M, Sun X, Li G. Resveratrol Alleviates Inflammation and ER Stress Through SIRT1/NRF2 to Delay Ovarian Aging in a Short-Lived Fish. The journals of gerontology Series A, Biological sciences and medical sciences. 2023;78:596-602
158. Guerrero-Netro HM, Estienne A, Chorfi Y, Price CA. The mycotoxin metabolite deepoxy- deoxynivalenol increases apoptosis and decreases steroidogenesis in bovine ovarian theca cells. Biology of reproduction. 2017;97:746-57
159. Takahashi N, Harada M, Hirota Y, Nose E, Azhary JM, Koike H. et al. Activation of Endoplasmic Reticulum Stress in Granulosa Cells from Patients with Polycystic Ovary Syndrome Contributes to Ovarian Fibrosis. Scientific reports. 2017;7:10824
160. Zhang Y, Chen X, Lin Y, Liu X, Xiong X. Identification of crucial pathways and genes linked to endoplasmic reticulum stress in PCOS through combined bioinformatic analysis. Frontiers in molecular biosciences. 2024;11:1504015
161. Xiang Y, Wang H, Ding H, Xu T, Liu X, Huang Z. et al. Hyperandrogenism drives ovarian inflammation and pyroptosis: A possible pathogenesis of PCOS follicular dysplasia. International immunopharmacology. 2023;125:111141
162. Jabarpour M, Aleyasin A, Nashtaei MS, Lotfi S, Amidi F. Astaxanthin treatment ameliorates ER stress in polycystic ovary syndrome patients: a randomized clinical trial. Scientific reports. 2023;13:3376
163. Jabarpour M, Amidi F, Aleyasin A, Nashtaei MS, Marghmaleki MS. Randomized clinical trial of astaxanthin supplement on serum inflammatory markers and ER stress-apoptosis gene expression in PBMCs of women with PCOS. Journal of cellular and molecular medicine. 2024;28:e18464
164. Brenjian S, Moini A, Yamini N, Kashani L, Faridmojtahedi M, Bahramrezaie M. et al. Resveratrol treatment in patients with polycystic ovary syndrome decreased pro-inflammatory and endoplasmic reticulum stress markers. American journal of reproductive immunology (New York, NY: 1989). 2020;83:e13186
165. El-Saka MH, Barhoma RA, Ibrahim RR, Elsaadany A, Alghazaly GM, Elshwaikh S. et al. Potential effect of adrenomedullin on metabolic and endocrinal dysfunctions in the experimentally induced polycystic ovary: Targeting implication of endoplasmic reticulum stress. Journal of biochemical and molecular toxicology. 2021;35:e22725
166. Bai Y, Liu Y, Wang Y, Liu X, Wang Y, Liu H. et al. IL-18BP Therapy Ameliorates Reproductive and Metabolic Phenotypes in a PCOS Mouse Model by Relieving Inflammation, Fibrosis and Endoplasmic Reticulum Stress. Reproductive sciences (Thousand Oaks, Calif). 2024;31:3595-608
167. Peng Y, Guo L, Gu A, Shi B, Ren Y, Cong J. et al. Electroacupuncture alleviates polycystic ovary syndrome-like symptoms through improving insulin resistance, mitochondrial dysfunction, and endoplasmic reticulum stress via enhancing autophagy in rats. Molecular medicine (Cambridge, Mass). 2020;26:73
168. Cong J, Zhang Y, Yang X, Wang Y, He H, Wang M. Anti-polycystic ovary syndrome effect of electroacupuncture: IMD inhibits ER stress-mediated apoptosis and autophagy in granulosa cells. Biochemical and biophysical research communications. 2022;634:159-67
169. Cozzolino M, Herraiz S, Cakiroglu Y, Garcia-Velasco JA, Tiras B, Pacheco A. et al. Distress response in granulosa cells of women affected by PCOS with or without insulin resistance. Endocrine. 2023;79:200-7
170. Bañuls C, Rovira-Llopis S, Martinez de Marañon A, Veses S, Jover A, Gomez M. et al. Metabolic syndrome enhances endoplasmic reticulum, oxidative stress and leukocyte-endothelium interactions in PCOS. Metabolism: clinical and experimental. 2017;71:153-62
171. Chiang YF, Lin IC, Huang KC, Chen HY, Ali M, Huang YJ. et al. Caffeic acid's role in mitigating polycystic ovary syndrome by countering apoptosis and ER stress triggered by oxidative stress. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2023;166:115327
172. Zhang W, Wu F. Linoleic acid induces human ovarian granulosa cell inflammation and apoptosis through the ER-FOXO1-ROS-NFκB pathway. Scientific reports. 2024;14:6392
173. He Q, Qu M, Shen T, Su J, Xu Y, Xu C. et al. Control of mitochondria-associated endoplasmic reticulum membranes by protein S-palmitoylation: Novel therapeutic targets for neurodegenerative diseases. Ageing research reviews. 2023;87:101920
174. Pedriali G, Rimessi A, Sbano L, Giorgi C, Wieckowski MR, Previati M. et al. Regulation of Endoplasmic Reticulum-Mitochondria Ca(2+) Transfer and Its Importance for Anti-Cancer Therapies. Frontiers in oncology. 2017;7:180
175. Yuan C, Huang WQ, Guo JH, Liu XY, Yang JZ, Chen JJ. et al. Involvement of kisspeptin in androgen-induced hypothalamic endoplasmic reticulum stress and its rescuing effect in PCOS rats. Biochimica et biophysica acta Molecular basis of disease. 2021;1867:166242
176. Lan B, He Y, Sun H, Zheng X, Gao Y, Li N. The roles of mitochondria-associated membranes in mitochondrial quality control under endoplasmic reticulum stress. Life sciences. 2019;231:116587
177. Ding H, Xiang Y, Zhu Q, Wu H, Xu T, Huang Z. et al. Endoplasmic reticulum stress-mediated ferroptosis in granulosa cells contributes to follicular dysfunction of polycystic ovary syndrome driven by hyperandrogenism. Reproductive biomedicine online. 2024;49:104078
178. Li X, Lin Q, Gou F, Zhu J, Yu M, Hong Q. et al. Effects of hesperidin on mitochondrial function, mitochondria-associated endoplasmic reticulum membranes and IP3R-MCU calcium axis in the intestine of piglets exposed to deoxynivalenol. Food & function. 2024;15:6459-74
179. Chang H, Yang F, Bai H, Lu Z, Xing C, Dai X. et al. Molybdenum and/or cadmium induce NLRP3 inflammasome production by causing mitochondria-associated endoplasmic reticulum membrane dysfunction in sheep hepatocytes. Chemico-biological interactions. 2023;382:110617
180. Joshi A. PCOS stratification for precision diagnostics and treatment. Frontiers in cell and developmental biology. 2024;12:1358755
181. Qasemi M, Aleyasin A, Mahdian R, Ghanami Gashti N, Shabani Nashtaei M, Ashrafnezhad Z. et al. Cell-free mtDNA level and its biomarker potency for ART outcome are different in follicular fluid of PCOS and non-PCOS women. Mitochondrion. 2021;59:30-6
182. Gongadashetti K, Gupta P, Dada R, Malhotra N. Follicular fluid oxidative stress biomarkers and ART outcomes in PCOS women undergoing in vitro fertilization: A cross-sectional study. International journal of reproductive biomedicine. 2021;19:449-56
Author contact
![]() Corresponding author: Jichun Tan. Email: tanjcorg. Liu Cao. Email: lcaoedu.cn.
Corresponding author: Jichun Tan. Email: tanjcorg. Liu Cao. Email: lcaoedu.cn.