Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Molecular Machinery of...
3. Selective Autophagy: A...
4. Disease-Specific Mechanisms
5. Therapeutic Strategies
6. Conclusion and Perspectives
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(11):6132-6148. doi:10.7150/ijbs.127431 This issue Cite
Review
Deciphering the Neuroautophagic Interactome: Molecular Circuits Linking Selective Autophagy to Neuropathological Cascades in Neurological Disorders
Pengfei Luo1,2,3, Zachary D. Travis4, Cameron Lenahan5, Haijian Wu1,2, Jianmin Zhang1,2, Jun Yu1,2, ![]() , Weilin Xu1,2,3,
, Weilin Xu1,2,3, ![]()
1. Department of Neurosurgery, Second Affiliated Hospital, School of Medicine, Zhejiang University, 88 Jiefang Rd, Hangzhou, Zhejiang 310009, China.
2. Zhejiang Key Laboratory of Research and Transformation for Major Neurosurgical Diseases, Hangzhou 310009, China.
3. State Key Laboratory of Transvascular Implantation Devices, Hangzhou 310009, China.
4. Department of Medical Sciences, College of Health Sciences, Western University of Health Sciences, Pomona, Ca, USA.
5. Burrell College of Osteopathic Medicine at New Mexico State University, Las Cruces, NM, USA.
Received 2025-10-28; Accepted 2026-5-18; Published 2026-6-10
Abstract

Selective autophagy, a lysosome-dependent degradation pathway targeting specific substrates (e.g., mitochondria, protein aggregates), plays a pivotal role in maintaining neuronal homeostasis. Its dysregulation is intricately linked to neurodegenerative diseases, acute brain injuries, and neuroinflammatory disorders. This review elucidates the crosstalk between selective autophagy and key neuropathophysiological processes, including apoptosis, neuroinflammation, oxidative stress, and blood-brain barrier disruption. We delineate the dual roles of selective autophagy through the framework of the neuroautophagic interactome—a network in which kinases (ULK1, TBK1) and effectors (PINK1/Parkin, SQSTM1/p62) collaboratively interpret ubiquitin codes. This integrated signaling nexus functions as a decisive hub that bidirectionally modulates disease progression. Furthermore, we evaluate emerging therapeutic strategies targeting selective autophagy to mitigate neuronal damage, emphasizing its dual role as both a protector and a contributor to disease progression.
Keywords: selective autophagy, neuroautophagic interactome, neurological disorders, therapeutic targets
1. Introduction
Selective autophagy, a precise degradation system that directly maintains neuronal homeostasis by clearing dysfunctional mitochondria (mitophagy) and toxic protein aggregates (aggrephagy), governs neuronal survival and proteostasis. Additionally, it modulates apoptosis, neuroinflammation, oxidative stress, BBB integrity, and proteinopathy in neurological disorders (1,2). We propose the neuroautophagic interactome, defined as a dynamic, bidirectional regulatory network that integrates the autophagy-lysosomal machinery with the pathophysiological processes of neurological diseases. Transcending conventional linear degradation models, this framework elucidates the reciprocal interactions between molecular nodes—including autophagy receptors (e.g., p62, OPTN), ubiquitination machinery (e.g., Parkin, LUBAC), regulatory kinases (e.g., ULK1, TBK1), and functional edges in signaling and trafficking that undergo disease-specific “rewiring” (3,4). In Alzheimer's disease (AD), for instance, impaired lysosomal clearance converts autophagy into a neurotoxic driver (5), whereas in acute injuries like stroke, transient mitophagy protects penumbral neurons before transitioning to ferroptosis (6). By mapping these systemic feedback loops and decoding the interactome's molecular logic, we can identify critical hub nodes and inform stage-specific therapeutic strategies, such as nanocarrier-mediated TFEB delivery to restore proteostasis (7). Biomarkers like CSF LC3-II/p62 ratio may predict clinical outcomes (8). Here, by decoding the interactome's molecular logic, we bridge organelle-level mechanisms and circuit dysfunction, redefining autophagy as a central hub in neurological pathogenesis and therapy.
2. Molecular Machinery of Selective Autophagy
The exquisite precision of selective autophagy in neural cells arises from a tightly coordinated molecular machinery, comprising four interconnected systems: autophagy-related (ATG) proteins, cargo receptors, ubiquitination machinery, and regulatory kinases. These components collectively enable the recognition, engulfment, and degradation of specific substrates, from damaged mitochondria to pathogenic protein aggregates, while maintaining metabolic and proteostatic equilibrium in the nervous system (Figure 1).
Molecular machinery of selective autophagy. Autophagy initiation is triggered by the ULK1/ATG1 kinase complex (ULK1- ATG13-FIP200-ATG101), which activates the class III PI3K complex I (VPS34-BECN1-ATG14L) to nucleate a phagophore at ER-mitochondria contact sites. Membrane expansion is then driven by two systems: (i) the ATG12-ATG5-ATG16L1 complex that promotes LC3/GABARAP lipidation, and (ii) ATG9 vesicles that deliver lipids from recycling endosomes. As the phagophore matures, selective autophagy receptors (p62/SQSTM1, OPTN, NBR1, NDP52) are recruited. Concurrently, misfolded proteins, soluble oligomers, and damaged mitochondria undergo ubiquitination through the collaboration of K48 and K63 linkages, mediated by CHIP, Parkin, and LUBAC. Ubiquitinated cargo is captured by receptor-decorated phagophores and subsequently cleared through lysosomal degradation. This process is regulated by ULK1, AMPK, mTORC1, and TBK1.
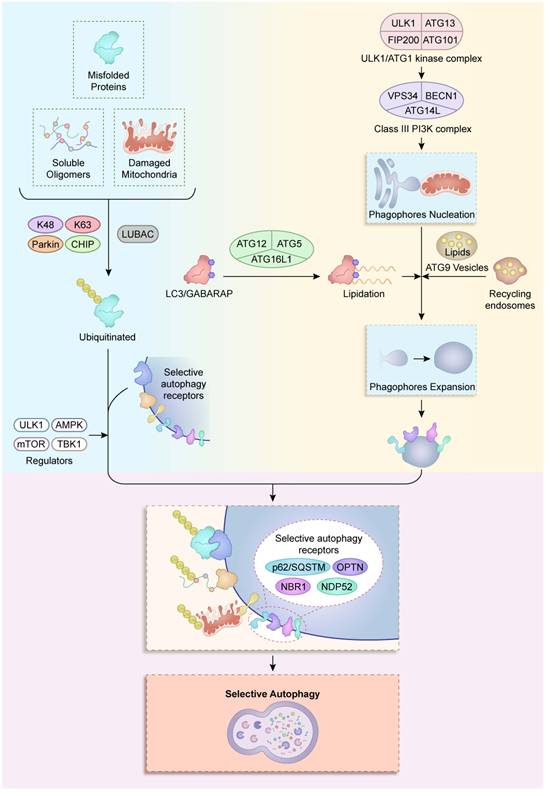
2.1 Core Autophagy-Related Proteins (ATG Proteins)
Central to all autophagy pathways are the evolutionarily conserved ATG proteins, which execute autophagosome formation through hierarchical assembly. The ULK1/ATG1 kinase complex (ULK1-ATG13-FIP200-ATG101) acts as the initiation switch by integrating nutrient signals via mTORC1 and AMPK-dependent phosphorylation (9). In neurons, ULK1 exhibits compartment-specific regulation. Specifically, dendritic ULK1 is inhibited by NMDA-receptor overactivation during excitotoxicity, whereas axonal ULK1 pools are activated by local ATP depletion (10,11). Downstream of this initiation, the class III PI3K complex (VPS34-BECN1-ATG14L) catalyzes the production of phosphatidylinositol-3-phosphate (PI3P), which subsequently recruits WIPI2b and DFCP1 to nucleate phagophores at endoplasmic reticulum (ER)-mitochondria contact sites (12). Furthermore, these pathways undergo neural-specific adaptations; for instance, BECN1 plays a bifurcated role in both canonical autophagosome biogenesis and the specialized pruning of GABAergic synapses via LC3-associated endocytosis (13,14).
Membrane expansion is driven by two ubiquitin-like conjugation systems: the ATG12-ATG5-ATG16L1 complex facilitates LC3/GABARAP lipidation (15), whereas ATG9 vesicles shuttle lipids from recycling endosomes. This latter process is disrupted in Huntington's disease by mutant huntingtin binding to ATG9A's C-terminal domain (16,17). Notably, LC3 isoforms show functional divergence: GABARAPL1 preferentially binds mitochondrial prohibitin-2 during mitophagy, whereas LC3B is essential for synaptic vesicle autophagy (18). Neurons further employ unique ATG protein paralogs; for instance, cortical neurons express an ATG4B splice variant resistant to oxidative inactivation, thereby ensuring autophagic flux under high ROS conditions (19,20).
2.2 Selective Autophagy Receptors (p62/SQSTM1, OPTN, NBR1, NDP52)
Selective autophagy receptors serve as molecular interpreters that bridge ubiquitinated cargo to the autophagic machinery. p62/SQSTM1 oligomerizes via its N-terminal PB1 domain, clustering polyubiquitinated substrates (e.g., tau oligomers) into aggresomes, while its LIR motif recruits LC3-decorated membranes (21). In amyotrophic lateral sclerosis (ALS), mutant SOD1 disrupts p62's UBA domain, leading to defective aggregate clearance (22). OPTN (optineurin) exhibits dual mitophagy and immune functions under the regulation of TBK1 phosphorylation at Ser177. Its E478G mutation impairs both Parkin recruitment and IFN-β production in microglia (23). NBR1's J-domain enables chaperone-mediated extraction of aggregated proteins from neuronal nucleoli, while NDP52's SKICH domain targets cytosolic pathogens in astrocytes through TRIM21 collaboration (24).
Receptor redundancy and competition define neural selectivity: dopaminergic neurons prioritize OPTN for mitochondrial quality control, whereas oligodendrocytes rely on NBR1 for myelin debris clearance (25). Phase separation dynamics further regulate receptor activity as p62 undergoes liquid-liquid phase separation (LLPS) with K63-linked ubiquitin chains under oxidative stress. This process forms dynamic condensates that enhance aggrephagy efficiency (26). However, in FTLD-TDP patients, TDP-43 inclusions sequester p62's PB1 domain, which subsequently stalls condensate formation and promotes proteostatic collapse (27).
2.3 Ubiquitination Systems (Parkin, CHIP, LUBAC)
Ubiquitin codes dictate cargo specificity through E3 ligase-substrate pairing. Parkin, an RBR E3 ligase activated by PINK1-mediated phospho-ubiquitin signaling, tags damaged mitochondria with K63/K27 polyubiquitin chains for OPTN/NDP52 recognition. Importantly, this process requires neuronal calcium flux for full activation (28). CHIP (STUB1), in complex with HSP70, triages misfolded proteins (α-synuclein, TDP-43) to either proteasomal or autophagic degradation via substrate-specific ubiquitination: K48-linked chains target soluble oligomers to proteasomes, whereas K63 chains direct aggregates to autophagosomes (29). In Parkinson's disease, CHIP's T246M mutation biases ubiquitination toward K48 linkages, thereby exacerbating proteasomal overload (30).
The linear ubiquitin chain assembly complex (LUBAC; HOIP/HOIL-1/Sharpin) installs Met1-linked chains that stabilize autophagy-inflammation crosstalk (31). HOIP-generated linear ubiquitin on ASC specks recruits NDP52 to suppress NLRP3 inflammasomes in microglia, a checkpoint that is often disrupted in multiple sclerosis (MS) lesions (32). Paradoxically, LUBAC also enables mitophagy by linear ubiquitination of SMAC/DIABLO, preventing caspase-9 activation during mitochondrial stress (33). Such duality underscores ubiquitination's role as a rheostat balancing autophagy's cytoprotective and inflammatory outputs.
2.4 Regulatory Kinases (ULK1, AMPK, mTOR, TBK1)
A kinase network orchestrates autophagy dynamics across neural sub-compartments. ULK1's spatial regulation is critical: synaptic ULK1 is activated by CaMKK2 during long-term potentiation to clear aged mitochondria, while somato-dendritic ULK1 responds to AMPK-mediated glucose deprivation (34,35). AMPK itself exhibits isoform-specific roles. For instance, AMPKα2 has been reported to stabilize lysosomal V-ATPase in axons, whereas AMPKα1 promotes mitophagy via DRP1 phosphorylation (36). mTORC1's lysosomal positioning via RAGC GTPases creates subcellular autophagy mosaicism; in Alzheimer's neurons, mTOR hyperactivation at dendritic lysosomes blocks Aβ degradation but enhances Tau clearance via compartment-specific TFEB regulation (36).
TBK1 emerges as a linchpin connecting autophagy to neuroimmunity. Biochemical studies indicate that by phosphorylating OPTN (Ser177) and p62 (Ser403), TBK1 enhances their LC3-binding affinities ~20-fold, while concurrently phosphorylating STING to suppress neurotoxic type-I IFN responses (37). ALS-linked TBK1 mutations (e.g., E696K) decouple these functions by preserving OPTN phosphorylation and abolishing STING suppression, which would explain the concomitant autophagy failure and unchecked neuroinflammation observed in those cases (37). Kinase cross-talk is exemplified in ischemic stroke: HIF-1α-induced BNIP3 displaces BECN1 from BCL-2 to unleash autophagy, while the AMPK-mTOR-TBK1 axis dictates the ultimate “rebooting” capacity of the lysosomal system during reperfusion (38,39).
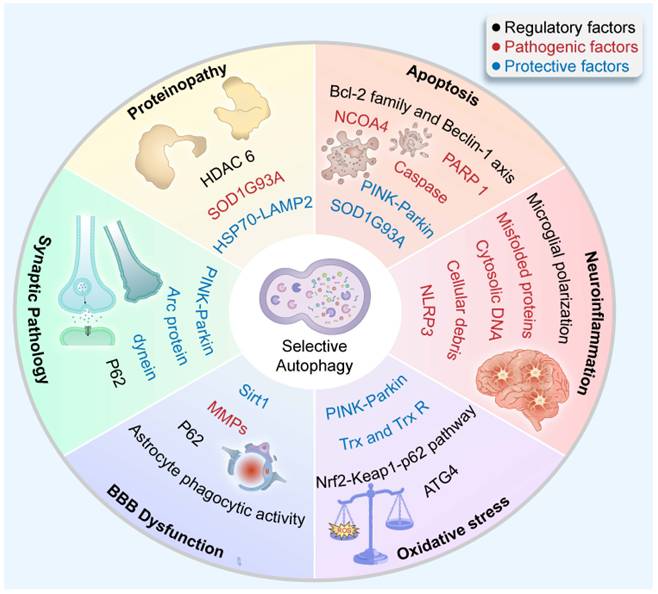
3. Selective Autophagy: A Multidimensional Interactome Bridging Cellular Stress Responses (Figure 2)
3.1 Selective Autophagy and Apoptosis: Molecular Switches Governing Survival and Demise
The interplay between selective autophagy and apoptosis in neurological disorders constitutes a delicate equilibrium, where molecular switches determine cellular fate. Central to this crosstalk is the Bcl-2 family/Beclin-1 axis, which serves as a rheostat balancing survival and death (40). In the context of Alzheimer's disease (AD), Aβ oligomers disrupt this axis, leading to excessive mitophagy that depletes neuronal ATP reserves (41-43). Conversely, in Parkinson's disease (PD), stabilized Bcl-2/Beclin-1 binding can suppress autophagy while simultaneously promoting BAX-mediated dopaminergic neuron apoptosis (44). This duality is exploited therapeutically; for instance, the BH3 mimetic ABT-737 disrupts Bcl-2/Beclin-1 complexes in glioblastoma, restoring autophagic flux while synergizing with temozolomide-induced apoptosis (45).
Selective Autophagy: A Multidimensional Interactome Bridging Cellular Stress Responses. Within the neuroautophagic interactome, selective autophagy exerts context-dependent effects—beneficial or detrimental—across diverse cellular stress responses in disease.
Caspase-mediated cleavage of autophagy proteins further remodels this interactome. During glutamate excitotoxicity in Huntington's disease, caspase-cleaved Bcl-2 fragments amplify mitochondrial damage (46), while caspase-8-mediated processing of SQSTM1/p62 impairs the clearance of Tau aggregates in AD neurons (47). Notably, caspase-6 exhibits paradoxical roles: its cleavage of VPS34 at Asp249 disrupts autophagosome formation in ALS motor neurons (48), while processing OPTN at Asp147 enhances OPTN's LC3 binding to sustain mitophagy in early-stage PD (49).
Beyond caspase regulation, the PINK1/Parkin axis orchestrates a mitochondrial control system. Under physiological stress, PINK1 is stabilized on depolarized mitochondria, recruiting Parkin to ubiquitinate VDAC1 with K27/K63 chains for OPTN-mediated mitophagy (50). However, sustained damage switches Parkin's activity. Specifically, in PD-linked Parkin (R275W) mutants, impaired ubiquitination leads to BAD (Bcl-2 associated agonist of cell death), opening mitochondrial permeability transition pores and triggering caspase-9 apoptosis (51). This bidirectional regulation is spatially encoded—synaptic mitochondria rely on PINK1/Parkin for routine turnover, while somatic mitochondria employ BCL2L13 for Parkin-independent mitophagy (52). Furthermore, α-synuclein mutants subvert this system by sequestering Parkin, coupling proteostatic collapse with increased apoptotic susceptibility (53,54).
In demyelinating disorders like MS, the interactome bridges ferroptosis-lipophagy crosstalk. Iron-loaded microglia undergo ferroptotic death via ACSL4-mediated lipid peroxidation, releasing oxidized phospholipids that activate astrocytic lipophagy through PPARγ-TFEB signaling (55). This response was proposed as a means to clear toxic lipid droplets but it depletes plasmalogens essential for myelin integrity. The intersection is regulated by NCOA4: during remyelination, NCOA4 mediates ferritinophagy to buffer iron, while its oxidation by 4-HNE in chronic MS redirects it to degrade lipidated LC3, stalling autophagic flux (56,57). Therapeutic modulation of this axis shows promise, liproxstatin-1, a ferroptosis inhibitor, enhances remyelination via NRF2-driven lipophagy upregulation (58).
In patients with ALS, SOD1G93A mutants induce IRE1α activation, which simultaneously cleaves Bcl-2 mRNA (reducing autophagy) and activates apoptosis (59). Moreover, ischemia-reperfusion injury induces HMGB1-dependent PARP1 hyperactivation, generating PAR polymers that block the p62-ZZ domain from binding ubiquitin, stalling aggrephagy while inducing caspase-independent apoptosis (60).
Mitochondrial injury and proteotoxicity act as decisive inputs that reshape the interactome's topology. By modulating molecular rheostats like the Bcl-2/Beclin-1 complex and PINK1/Parkin axis, these stressors force a critical transition: the interactome shifts from a homeostatic "survival mode" (promoting cargo degradation) to a "demise mode" (triggering apoptotic cascades), effectively determining the point of no return for neuronal viability.
3.2 Selective Autophagy and Neuroinflammation: Bidirectional Regulation of Immunity and Degradation
Neuroinflammation is a critical component in the pathogenesis of various neurological disorders, including AD, PD, MS, and ALS. It involves the activation of glial cells, particularly microglia and astrocytes, leading to the release of pro-inflammatory cytokines, chemokines, and reactive oxygen species (ROS). Selective autophagy has emerged as a key regulatory mechanism in modulating neuroinflammatory responses, offering potential therapeutic targets for these debilitating conditions (61).
The interplay between selective autophagy and neuroinflammation is complex and bidirectional. One of the most studied pathways involves the NLRP3 inflammasome, a multiprotein complex that drives the maturation of interleukin-1β (IL-1β) and IL-18. Selective autophagy negatively regulates NLRP3 inflammasome activation by degrading damaged mitochondria, a major source of mitochondrial ROS (mtROS) and mitochondrial DNA (mtDNA), which act as danger-associated molecular patterns (DAMPs) (62). In neurodegenerative diseases like AD and PD, impaired mitophagy leads to the accumulation of damaged mitochondria, perpetuating NLRP3 inflammasome activation and chronic neuroinflammation (63,64).
Moreover, selective autophagy plays a crucial role in the clearance of other DAMPs, including misfolded proteins and cellular debris, which are potent triggers of neuroinflammatory responses. For example, in ALS, the accumulation of TDP-43 aggregates activates microglia and astrocytes, exacerbating neuroinflammation (65). Autophagy pathways such as aggrephagy are essential for the degradation of these toxic protein aggregates, thereby mitigating neuroinflammatory cascades (66,67).
Parallel to proteinopathy, the STING (stimulator of interferon genes) pathway, activated by cytosolic DNA, also intersects with selective autophagy. Evidence has shown that in MS, impaired autophagy results in the accumulation of cytosolic DNA, leading to STING pathway activation and the production of type I interferons, which drive neuroinflammation and demyelination (68,69). Enhancing selective autophagy could attenuate STING-mediated neuroinflammation in MS and other neuroinflammatory disorders (69,70).
Microglial polarization, a process by which microglia switch between pro-inflammatory (M1) and anti-inflammatory (M2) phenotypes, is another interface where selective autophagy exerts control. In AD, for instance, impaired autophagy in microglia promotes a sustained M1 phenotype characterized by the release of pro-inflammatory cytokines and neurotoxic factors. Conversely, autophagy induction has been shown to promote microglial polarization toward the M2 phenotype, facilitating tissue repair and anti-inflammatory responses (71,72).
Taken together, these mechanisms illustrate how the neuroautophagic interactome is rewired in response to DAMPs and cytosolic DNA. Under inflammatory stress, the network's priority shifts from routine proteostasis to immunological regulation, where autophagic hubs serve as negative feedback loops to quench inflammasome hyperactivation and prevent a feed-forward cycle of neuroinflammation.
3.3 Selective Autophagy and Oxidative Stress: Redox Homeostasis Through Organelle Crosstalk
Oxidative stress is a hallmark of numerous neurological disorders, including AD, PD, ALS, and stroke. It arises from an imbalance between the production of ROS and the capacity of cellular antioxidant defenses to neutralize them (73). Excessive ROS can damage lipids, proteins, and DNA, leading to neuronal dysfunction and death. Selective autophagy, particularly mitophagy, plays a pivotal role in mitigating oxidative stress by maintaining mitochondrial quality and redox homeostasis, thus offering a potential therapeutic avenue for these diseases (74,75).
The Nrf2-Keap1-p62 pathway is a key regulatory axis that links selective autophagy to oxidative stress (76). Under conditions of oxidative stress, p62/SQSTM1 (a selective autophagy receptor) accumulates and competitively binds to Keap1, freeing Nrf2 to translocate into the nucleus. Nrf2 then activates the transcription of antioxidant genes such as HO-1 and NQO1, bolstering cellular defenses against ROS (77). In neurodegenerative disorders like AD and PD, impaired autophagy leads to the accumulation of damaged proteins and organelles, further exacerbating oxidative stress (78). Enhancing selective autophagy, particularly through the p62-Nrf2 axis, could thus bolster antioxidant responses and mitigate neuronal damage (79).
Mitophagy, the selective degradation of damaged mitochondria, is critical for reducing oxidative stress. Dysfunctional mitochondria are a major source of ROS, and their accumulation is a common feature in neurological diseases (80). For instance, in PD, mutations in PINK1 and Parkin disrupt mitophagy, leading to the accumulation of defective mitochondria and increased ROS production (81). Similarly, in ALS, impaired mitophagy contributes to mitochondrial dysfunction and oxidative stress, driving motor neuron degeneration (82,83). Therapeutic strategies aimed at enhancing mitophagy could therefore alleviate oxidative stress and improve neuronal survival in these disorders.
ROS also directly regulate autophagy at the molecular level. For instance, evidence suggests that ROS may modulate the activity of ATG4, a cysteine protease essential for processing LC3 (a key autophagy protein). Oxidative modifications of ATG4 appear to either inhibit or activate its function, depending on the extent and context of ROS exposure (84). This regulatory mechanism ensures that autophagy is appropriately tuned to the cellular redox state, preventing excessive or insufficient autophagic activity. In stroke, where oxidative stress is a major driver of neuronal injury, fine-tuning autophagy through AMPK modulation could offer protection against ischemia-reperfusion damage (85).
The thioredoxin system, which includes thioredoxin (Trx) and thioredoxin reductase (TrxR), is another critical component of the cellular antioxidant machinery that interacts with selective autophagy (86). Trx regulates redox-sensitive proteins, including those involved in autophagy, and its activity is often compromised in neurological diseases (87,88). In ALS, for example, mutations in SOD1 disrupt the thioredoxin system, leading to increased oxidative stress and impaired autophagy (89,90). Enhancing the thioredoxin system or targeting its interplay with selective autophagy could thus provide therapeutic benefits in ALS and other oxidative stress-related disorders.
Oxidative stress fundamentally reshapes the neuroautophagic interactome by focusing its activity on redox-sensitive molecular hubs. Specifically, the ROS-induced activation of the Nrf2-Keap1-p62 axis and the fine-tuning of ATG4 activity redirect the autophagic machinery toward mitochondrial quality control and antioxidant defense, establishing a defensive "redox-buffering" signature within the network.
3.4 Selective Autophagy and Blood-Brain Barrier Dysfunction: Dynamic Remodeling of Neurovascular Integrity
Within this complex pathological landscape, the blood-brain barrier (BBB), composed of specialized endothelial cells, pericytes, astrocytes, and tight junction proteins, serves as a dynamic interface regulating molecular exchange between the systemic circulation and the central nervous system (CNS). Emerging evidence demonstrates that the BBB is dynamically regulated by selective autophagy in endothelial cells and astrocytes (91-93). Under physiological conditions, dysfunctional or misfolded tight junction proteins (e.g., claudin-5, ZO-1) are selectively targeted for lysosomal degradation via p62-mediated aggrephagy, preventing paracellular leakage (maintaining BBB integrity) (94). However, this homeostatic process is subverted during acute injury. In ischemic stroke, evidence suggests that hypoxia-inducible factor-1α (HIF-1α) upregulates BNIP3, which triggers excessive autophagy that degrades occludin and exacerbates BBB disruption (92,95). Concurrently, damaged endothelial mitochondria release mtDNA, activating STING-dependent neuroinflammation, while selective mitophagy fails to compensate due to Parkin downregulation (68,96-100). Astrocytic end-feet may clear pathological deposits (e.g., fibrinogen) through phagocytic activity, but chronic activation could promote protease secretion (e.g., MMP-9), contributing to basement membrane degradation (101-104).
Therapeutic strategies to protect BBB integrity have shown promise: ULK1 activators (e.g., LYN-1604) have been reported to restore autophagic flux and reduce edema, and nanoparticle-mediated TFEB delivery shows promise in enhancing lysosomal clearance of hemoglobin degradation products in subarachnoid hemorrhage (105-107).
Within the neurovascular unit, stressors such as hypoxia and metabolic failure convert the neuroautophagic interactome from a stabilizer of tight junction integrity into a mediator of barrier breakdown. This transition involves a fundamental displacement of the interactome's core nodes, illustrating how external vascular stress can subvert autophagic flux to compromise the structural stability of the blood-brain barrier.
3.5 Selective Autophagy and Synaptic Pathology: Quality Control at the Neural Circuit Level
Parallel to the regulatory processes at the structural barriers, synaptic dysfunction and degeneration are pivotal events in the pathogenesis of numerous neurological and psychiatric disorders, reflecting the critical role of synaptic plasticity in maintaining neural network integrity. Emerging evidence highlights synaptic pathology as an early biomarker of neurodegenerative processes, often preceding overt neuronal loss.
Synaptic integrity relies on spatially constrained selective autophagy to maintain proteostasis and mitochondrial fitness at nerve terminals (108). Synaptic vesicles and aged mitochondria are preferentially degraded via local (synaptic) autophagy, a process requiring local translation of PINK1 mRNA and dynein-mediated retrograde transport of autophagosomes (109,110). In Alzheimer's disease, tau pathology (e.g., hyperphosphorylated tau accumulation) is associated with impaired autophagy, potentially contributing to synaptic dysfunction (111-113). Similarly, in Parkinson's disease, α-synuclein aggregates disrupt autophagic clearance by promoting p62 oligomerization and lysosomal dysfunction (114,115). Emerging evidence suggests that the Arc-LC3C axis, essential for memory consolidation, electively degrades inactive AMPA receptors—a pathway subverted by protein inclusions in frontotemporal dementia (116,117). Pharmacological modulation of autophagy (e.g., exercise or rapamycin) shows therapeutic potential in preclinical models, though its precise effects on synaptic restoration require further investigation (118,119).
Collectively, synaptic stressors drive the interactome toward specialized 'synaptic signatures,' such as Arc-mediated internalization and retrograde transport, to protect circuits from early erosion.
3.6 Selective Autophagy and Proteinopathy: Combating Phase-Transitioned Pathogenic Aggregates
Proteinopathy, characterized by the accumulation of misfolded or aggregated proteins, is a unifying pathological feature across major neurodegenerative diseases. Selective autophagy plays a pivotal yet paradoxical role in this process, acting as both a protective clearance mechanism and a potential contributor to proteostatic collapse when dysregulated (120,121). The p62/SQSTM1 adapters facilitate sequestration of ubiquitinated aggregates into autophagosomes, a process enhanced by liquid-liquid phase separation (LLPS) (122,123). This mechanism is disrupted in ALS by SOD1G93A-induced oxidation of p62 (124,125). In addition to aggrephagy, HSP70-LAMP2A-reliant Chaperone-mediated autophagy (CMA), which selectively degrades soluble oligomers, is often overwhelmed in AD due to Aβ42-mediated suppression of LAMP2A transcription and ER stress-induced lysosomal dysfunction (126). HDAC6 further regulates the interactome by linking the UPS and autophagy pathways. Specifically, it recruits dynein motors to transport aggregates to the perinuclear “aggresome” for autophagic clearance—a pathway that is defective in Huntington's disease where mutant huntingtin sequesters HDAC6 (127,128). Emerging therapies like ATTEC compounds (autophagosome tethering compounds), which simultaneously bind LC3 and mutant huntingtin, enhance aggregate clearance in patient-derived neurons, while TFEB activators (e.g., curcumin analogs) restore proteostasis by amplifying lysosomal biogenesis and aggrephagy capacity (129).
Chronic proteotoxicity triggers a phase-transition from liquid-like degradation hubs to solid sequestration sites. This protective isolation, when overwhelmed, leads to the total proteostatic collapse seen in neurodegeneration.
3.7 Selective Autophagy and Cellular Heterogeneity: Mechanisms of Pathological Convergence
While selective autophagy is a conserved homeostatic mechanism, its operational priorities are highly cell-type specific within the CNS.
Neurons exhibit a highly specialized form of "compartmentalized" clearance necessitated by their extreme polarization. Unlike most cells, neuronal autophagosomes predominantly initiate at the distal axon and must undergo retrograde transport to the soma for lysosomal fusion (130). Evidence suggests that the "dying-back" neuropathy characteristic of PD and ALS is fundamentally a failure of this long-distance transport, where the inability to maintain distal axonal mitophagy leads to synaptic collapse long before somatic death (131,132).
In contrast, microglial responses are uniquely defined by the intersection of autophagy and phagocytic pathways, often described as LC3-associated phagocytosis (LAP) or LC3-associated endocytosis (LANDO). Recent findings indicate that microglia prioritize the clearance of extracellular aggregates, such as Aβ, through these non-canonical pathways. When these processes are impaired—often via TREM2 or ATG5 dysfunction—microglia shift from a neuroprotective phagocytic state toward a pro-inflammatory phenotype, triggering NLRP3 inflammasome activation and the release of neurotoxic cytokines (133,134). Furthermore, autophagic impairment causes microglia to propagate proteotoxic "seeds" (e.g., Tau fragments) via exosomes to healthy neurons, effectively transitioning these cells from homeostatic scavengers into pathological vectors of disease dissemination (135).
In a distinct role, astrocytes and endothelial cells prioritize the architectural maintenance of the neurovascular unit. Current progress highlights that astrocyte autophagy is indispensable for maintaining neural homeostasis and proteostasis, offering a promising therapeutic target for mitigating neurodegenerative progression (136). However, chronic overactivation in these cells can lead to the paradoxical degradation of tight junction proteins, such as occludin and claudin-5, thereby facilitating blood-brain barrier (BBB) breakdown in conditions like stroke and multiple sclerosis (93).
4. Disease-Specific Mechanisms
The molecular interplay between selective autophagy and neurological disorders manifests through disease-specific mechanisms that reflect both shared principles and unique pathophysiological adaptations. Below, we detail how autophagy contributes to or mitigates pathology in several major conditions.
4.1 Alzheimer's Disease (AD)
In AD, pathogenesis is marked by a progressive transition from targeted metabolic maintenance toward a global failure in molecular recognition, intersecting with proteinopathy and neuroinflammation to drive neuronal decline. Accumulating evidence suggests that Aβ42 oligomers interact with autophagic receptors such as p62/SQSTM1, impairing cargo recognition and promoting autophagosome accumulation; this process is further exacerbated by ATG16L1 dysfunction, which disrupts autophagosome maturation through VAMP7 mislocalization (137-139). Emerging evidence suggests that Aβ aggregates sequester GABARAP family proteins (e.g., GABARAPL1/2), blocking autophagosome-lysosome fusion and generating autophagic vesicles enriched with phosphorylated Tau, which act as nucleation sites for neurofibrillary tangle formation. This proteostatic collapse is further exacerbated by PSEN1-mediated lysosomal acidification defects (140) and Aβ42-induced ER stress, which suppresses chaperone-mediated autophagy (CMA) and allows toxic Tau oligomers to accumulate (141). The resulting persistent proteostatic failure activates microglial NLRP3 inflammasomes, a process driven by mitochondrial ROS leakage from mitophagy-deficient neurons. This mechanism creates a feed-forward loop of neuroinflammation and amyloidogenesis (142-144). Hippocampal neurons initially prioritize mitophagy to counteract OXPHOS failure. However, the depletion of metabolic substrates like NAD+ shifts the interactome from selective mitophagy to non-selective bulk degradation, accelerating neuronal demise (145).
4.2 Parkinson's Disease (PD)
In PD, pathogenesis revolves around α-synuclein's dual exploitation of autophagy and apoptosis pathways. The A53T mutant α-synuclein impairs chaperone-mediated autophagy (CMA) by blocking LAMP2A-dependent lysosomal uptake, leading to intracellular α-synuclein accumulation. LRRK2 mutations disrupt ATG9A vesicle trafficking and autophagosome formation in axons, which exacerbates protein aggregation in synaptic terminals (141,146-148). Pathologic α-synuclein propagates via exosome-mediated transfer and tunneling nanotubes, facilitated by LRP1-mediated endocytosis in recipient neurons, thereby spreading Lewy pathology (149-151). Dopaminergic neurons exhibit selective vulnerability due to compartmentalized autophagy failure: while somatic aggrephagy (via p62-LC3 interactions) initially compensates, chronic axonal autophagic flux impairment leads to synaptic degeneration and "dying-back" neuropathy. Paradoxically, while early caspase-mediated signaling may temporarily enhance mitophagy, chronic PINK1/Parkin dysfunction eventually forces a transition to mitochondrial-driven apoptosis (152-156). Iron accumulation in the substantia nigra exacerbates oxidative stress through NCOA4-ferritinophagy defects, while lipid peroxidation from impaired glutathione synthesis drives ferroptosis (56,157).
4.3 Multiple Sclerosis (MS)
In MS, the autophagic machinery is uniquely redirected from lipid homeostasis toward the active execution of neuroinflammatory cascades and blood-brain barrier disruption. Myelin-derived lipid peroxidation products (e.g., oxidized phospholipids) impair ULK1-mediated autophagosome initiation in oligodendrocyte precursor cells, leading to defective clearance of myelin debris and the activation of senescence-associated secretory phenotype (SASP) (158,159). Microglial lipophagy initially resolves inflammation through NBR1-dependent lipid droplet breakdown and fatty acid release. However, chronic activation redirects autophagy machinery to process gasdermin D, thereby promoting NLRP3 inflammasome assembly and pyroptosis (160-164). Genetic susceptibility in MS is linked to CLEC16A variants, which disrupt mitophagy in oligodendrocyte progenitors by impairing PINK1/Parkin-mediated mitochondrial clearance. This results in iron accumulation due to defective NCOA4-ferritinophagy, triggering ferroptotic cell death (165-167). BBB breakdown is amplified through two synergistic pathways: (1) HIF-1α-driven BNIP3 overexpression induces excessive endothelial autophagy that selectively degrades tight junction proteins (e.g., occludin), and (2) astrocytic LC3-associated phagocytosis of fibrinogen deposits paradoxically activates MMP-9 secretion via TLR4-NFκB signaling, leading to basement membrane degradation (92,168-171).
4.4 Amyotrophic Lateral Sclerosis (ALS)
In ALS, the autophagic system enters a state of functional exhaustion where the clearance machinery itself is physically sequestered by toxic aggregates, a process intertwined with TDP-43 proteinopathy and oxidative stress (172). Cytoplasmic TDP-43 aggregates evade clearance by disrupting interactions with HSP90 to impede proteasomal degradation and by promoting their sequestration into stress granules. Loss of nuclear TDP-43 compromises cryptic splicing repression (e.g., STMN2 cryptic exon inclusion), exacerbating neuronal dysfunction (172-174). The C9orf72 repeat expansion produces toxic dipeptide repeat proteins that subvert autophagy initiation. For example, poly-GA aggregates sequester ATG13 to disrupt ULK1 complexes, while poly-PR peptides hyperactivate RAB7A via USP10-mediated deubiquitination, causing lysosomal exhaustion (175). Furthermore, mutant SOD1 triggers pro-apoptotic ER stress and reduces autophagic flux via Bcl-2 cleavage, while concurrent p62 accumulation recruits RUNX2 to silence lysosomal genes (e.g., CTSD) (176-178). This proteostatic collapse is further compounded by the mTORC1-dependent sequestration of TFEB in the cytoplasm, which cripples lysosomal biogenesis. Pharmacological mTOR activation (e.g., PA treatment) partially restores TFEB nuclear localization and mitigates autophagic stress in TDP-43-deficient models (178-180).
4.5 Stroke
In stroke, the neuroautophagic interactome exhibits a sharp temporal duality, where the rapid transition from a neuroprotective scavenger to a mediator of vascular injury defines its role in BBB regulation and apoptosis. During the acute phase (0-6 h post-insult), HIF-1α-induced BNIP3/NIX mitophagy clears ROS-generating mitochondria, preserving penumbral neurons (181,182). However, reperfusion may trigger Parkin overexpression, diverting autophagosomes toward pathological mitochondrial fission rather than degradation (183). Delayed autophagy enhancement, such as administering rapamycin at 12 h post-stroke, exacerbates hemorrhagic transformation through MMP-9 activation. In contrast, circadian-aligned therapy capitalizes on morning autophagy peaks to extend the treatment window (184). Failed endothelial mitophagy releases mtDNA that activates STING-dependent neuroinflammation, which is compounded by FUNDC1-mitophagy insufficiency due to Parkin downregulation (185). The narrow therapeutic window highlights the need for spatiotemporal precision in autophagy-targeted interventions. ULK1 activators (e.g., LYN-1604 analogs) restore pericyte autophagic flux, mitigating vasogenic edema through mTORC1-TFEB axis modulation in stroke. Neutrophil-mimetic nanocarriers can deliver TFEB plasmids to ischemic microglia, enhancing lysosomal biogenesis and debris clearance (186-188).
These disease-specific paradigms underscore that selective autophagy is not a monolithic process, but rather a modular toolkit co-opted in various aspects of neurodegeneration. Therapeutic success hinges on decoding each disease's unique “autophagy signature”, the spatiotemporal balance between protective substrate clearance and pathological pathway hijacking. Advances in single-cell autophagy flux assays and conformation-specific autophagy modulators (e.g., Aβ fibril-targeting LIR peptides) promise to usher in an era of precision autophagy therapeutics tailored to specific neurological disease states.
5. Therapeutic Strategies
Building upon the molecular framework of autophagic regulation, diverse pharmacological interventions are being developed to restore cellular homeostasis in pathological states. The following sections evaluate these strategies (Table 1).
Potential Therapeutic Strategies in Neuroautophagy
| Therapeutic Treatments | Disease | Molecular Target | References | |
|---|---|---|---|---|
| Small Molecule Modulators | Rapamycin analogs | AD | mTORC1-FKBP12 | (189) |
| Spermidine | PD / stroke | PINK-Parkin / ASK p38 | (190,191) | |
| BL-918 | ALS | ULK | (193) | |
| mTOR inhibitors | epilepsy | HSP90α | (194,195) | |
| Gene Therapy | AAV9 | Batten disease | TEEB | (197,198) |
| miR-124-3p | PD | MEKK3-NFκB / p62/p38 axis | (199,200) | |
| CRISPRa-mediated ATG5 overexpression | MS | TREM2 | (201,202) | |
| miR-21 | TBI | mitochondrial clearance | (204,205) | |
| Nanocarrier Systems | PLGA nanoparticles functionalized with transferrin-receptor antibodies | AD | rapamycin accumulation | (206) |
| Lysosomal-targeting peptides (eg. GALA) | AD | PH-sensitive release of choroquine | (207,208) | |
| Neutrophil membrane-coated liposomes with TFEB plasmids | ischemic stroke | autophagy | (187,188) |
AD: Alzheimer's disease; PD: Parkinson's disease; ALS: Amyotrophic lateral sclerosis; MS: Multiple sclerosis; TBI: Traumatic brain injury
5.1 Small Molecule Modulators
Autophagy-targeted therapies require a careful balance between enhancing protective clearance and avoiding pathological overactivation. Rapamycin analogs (e.g., everolimus) enhance Aβ clearance in AD by disrupting mTORC1-FKBP12 interactions, but their immunosuppressive effects limit their use in MS (189). Spermidine exhibits dual neuroprotective effects: in Parkinson's disease (PD), it activates PINK1-Parkin-mediated mitophagy to clear dysfunctional mitochondria, whereas in stroke models, it reduces oxidative stress and preserves blood-brain barrier (BBB) integrity by modulating the ASK1-p38 pathway.
These effects are independent of the previously hypothesized CLDN5 acetylation mechanism (190-192). Third-generation compounds like BL-918 show context-dependent effects; in ALS, BL-918 activates ULK1 selectively in motor neurons via ROS-sensitive prodrug conversion while sparing inhibitory interneurons (193). However, off-target effects persist. For example, mTOR inhibitors can indirectly upregulate chaperone-mediated autophagy (CMA) by stabilizing HSP90α, as observed in epilepsy models (194-196).
5.2 Gene Therapy
Gene therapy approaches are being explored to modulate autophagy pathways. AAV9 vectors effectively target the central nervous system (CNS) by crossing the blood-brain barrier (BBB), enabling TFEB overexpression to enhance lysosomal biogenesis and restore cathepsin activity in lysosomal storage disorders such as Batten disease (197,198). miR-124-3p reduces α-synuclein propagation by suppressing neuroinflammation via MEKK3/NF-κB pathway inhibition while simultaneously enhancing chaperone-mediated autophagy (CMA) through p62/p38 axis modulation. This dual mechanism reduces Lewy body formation and preserves dopaminergic neurons (199,200). TREM2 activation (e.g., via AL002a) facilitates myelin debris clearance in MS models; alternatively, CRISPRa-mediated ATG5 overexpression is proposed as a potential strategy to enhance this process, though it may risk overactivating the inflammasome (201-203). Exosome-based therapies utilizing mesenchymal stem cell (MSC)-derived exosomes show promise in TBI. These exosomes deliver miR-21 and other neuroprotective miRNAs across the BBB, enhancing autophagy-mediated mitochondrial clearance and reducing secondary injury. Neutrophil-mimetic nanocarriers loaded with TFEB plasmids further improve lysosomal function in deep brain regions, overcoming light penetration limitations of optogenetic systems (204,205).
5.3 Nanocarrier Systems
Nanocarrier-based delivery systems offer targeted approaches to modulate autophagy in neurological diseases. For example, PLGA nanoparticles functionalized with transferrin-receptor antibodies increase rapamycin accumulation in the brain in AD models (206). Polymeric nanocarriers decorated with lysosomal-targeting peptides (e.g., GALA) enable pH-sensitive release of hydroxychloroquine in Aβ-rich regions, representing a promising strategy to enhance microglial Aβ clearance and potentially alleviate lysosomal dysfunction (207-209). Neutrophil membrane-coated liposomes can deliver TFEB plasmids to ischemic brain regions (achieving ~90% targeting efficiency in stroke) (187,188). However, long-term exposure to certain nanomaterials may have adverse effects. For instance, prolonged use of silica nanoparticles may induce ataxia in animal models via mitochondrial ROS overproduction and impaired lysosomal acidification (210,211).
6. Conclusion and Perspectives
The intricate interplay between selective autophagy and neurological disorders underscores autophagy's dual role as both a guardian and an executioner of neuronal homeostasis. This review, through the lens of the neuroautophagic interactome, highlights how autophagy machinery dynamically interfaces with apoptosis, neuroinflammation, oxidative stress, and other pathological cascades. These multifaceted interactions reveal context-dependent mechanisms that dictate disease progression. In neurodegenerative diseases such as AD and PD, impaired lysosomal clearance or subversion of autophagic pathways by pathogenic proteins (e.g., Tau, α-synuclein) exacerbates proteostatic collapse. In acute injuries like stroke, transient activation of BNIP3/NIX-mediated mitophagy clears ROS-generating mitochondria during ischemia, but reperfusion triggers excessive mitochondrial fission via Parkin-Drp1 hyperactivation, leading to lysosomal exhaustion and STING-dependent neuroinflammation.
Therapeutic strategies—including small-molecule autophagy modulators and nanocarrier-mediated delivery systems-demonstrate considerable promise. However, they face challenges in balancing efficacy with off-target effects, as demonstrated by rapamycin's immunosuppressive limitations via FKBP12-dependent T-cell inhibition and the narrow therapeutic windows of ULK1 activators (e.g., LYN-1604 analogs) due to compensatory CMA upregulation in non-target cells. Emerging technologies such as CRISPR-engineered autophagy reporters and AI-driven flux models hold potential to refine precision autophagy therapeutics, but their translation will require resolving critical gaps in spatial and temporal specificity.
Despite recent advances, several limitations persist. Preclinical models often fail to recapitulate human-specific autophagy adaptations (such as neuron-specific ATG4B splice variants or circadian-regulated flux dynamics), thus limiting translational relevance. The dual-edged nature of autophagy modulation remains a major hurdle: chronic autophagy induction risks lysosomal burnout, whereas autophagy inhibition may inadvertently amplify proteinopathy. Interventional targets are intimately connected with the autophagy state and cellular homeostasis as disease progresses. Hub regulators such as AMPK, mTORC1, and SIRT1 influence not only autophagy but also multiple processes implicated in neurodegenerative diseases, including epigenetic regulation and energy homeostasis (212). Organelle-targeted autophagy regulation, rather than global autophagy, underpins selective autophagy-based therapeutic strategies. The selective clearance of damaged components, while preserving normal cellular metabolism, could substantially widen the therapeutic window (129). Biomarkers like CSF LC3-II/p62 ratio, though informative, lack disease specificity, complicating clinical stratification. Technical barriers further hinder progress (8). For instance, current imaging tools cannot dynamically track autophagosome-lysosome interactions in vivo, and the off-target effects of systemically administered autophagy-modulating compounds (e.g., TFEB activators) remain poorly characterized.
Looking ahead, the field must prioritize strategies that recalibrate, rather than uniformly boost or suppress—autophagic activity. This demands innovations in cell-type-specific delivery systems, conformation-sensitive therapies targeting pathogenic aggregates, and closed-loop neuromodulation platforms responsive to metabolic cues. Collaborative efforts integrating structural biology, nanotechnology, and computational modeling will be essential to decode the spatiotemporal logic of the neuroautophagic interactome. Ultimately, bridging these gaps will not only illuminate autophagy's role in neurological pathogenesis but also pave the way for therapies that restore proteostatic resilience without causing collateral damage to the brain's fragile ecosystem.
Abbreviations
AD: Alzheimer's disease; ALS: Amyotrophic lateral sclerosis; BAD: Bcl-2 associated agonist of cell death; BBB: blood-brain barrier; CMA: Chaperone-mediated autophagy; CNS: central nervous system; DAMPs: danger-associated molecular patterns; LLPS: liquid-liquid phase separation; MS: Multiple sclerosis; MSC: mesenchymal stem cell; OPTN: optineurin; PD: Parkinson's disease; PI3P: phosphatidylinositol-3-phosphate; ROS: reactive oxygen species; TBI: Traumatic brain injury; Trx: thioredoxin; TrxR: thioredoxin reductase; UPS: ubiquitin-proteasome system.
Acknowledgements
The authors acknowledge the use of Deepseek for language polishing and grammatical editing to improve the clarity and flow of the entire manuscript. The authors have reviewed and edited the content generated by the tool and take full responsibility for the integrity and accuracy of the final manuscript.
Author contributions
P.L.: Writing - original draft, Methodology
Z.D.T., C.L.: Writing - review & editing
H.W., J. Z.: Supervision
J.Y., W.X.: Conceptualization, Writing - review & editing, Supervision.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Li W, He P, Huang Y, Li YF, Lu J, Li M. et al. Selective autophagy of intracellular organelles: recent research advances. Theranostics. 2021Jan1;11(1):222-56 doi:10.7150/thno.49860 PubMed PMID: 33391472; PubMed Central PMCID: PMC7681076
2. Scrivo A, Bourdenx M, Pampliega O, Cuervo AM. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018Sep;17(9):802-15 doi:10.1016/S1474-4422(18)30238-2 PubMed PMID: 30129476; PubMed Central PMCID: PMC6359907
3. Vargas JNS, Hamasaki M, Kawabata T, Youle RJ, Yoshimori T. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol. 2023Mar;24(3):167-85 doi:10.1038/s41580-022-00542-2
4. Lamark T, Johansen T. Mechanisms of Selective Autophagy. Annu Rev Cell Dev Biol. 2021Oct6;37:143-69 doi:10.1146/annurev-cellbio-120219-035530
5. Mary A, Eysert F, Checler F, Chami M. Mitophagy in Alzheimer's disease: Molecular defects and therapeutic approaches. Mol Psychiatry. 2023;28(1):202-16 doi:10.1038/s41380-022-01631-6 PubMed PMID: 35665766; PubMed Central PMCID: PMC9812780
6. Ma Y, Wang X, Li Y, Zhao J, Zhou X, Wang X. Mechanisms Associated with Mitophagy and Ferroptosis in Cerebral Ischemia-reperfusion Injury. J Integr Neurosci. 2025Mar18;24(3):26458 doi:10.31083/JIN26458
7. Liang X, Zeng Y, Zhang P, Zhu B, Feng J, Deng T. et al. Polystyrene nanoplastics trigger pyroptosis in dopaminergic neurons through TSC2/TFEB-mediated disruption of autophagosome-lysosome fusion in Parkinson's disease. J Transl Med. 2025Jun5;23:631 doi:10.1186/s12967-025-06634-9 PubMed PMID: 40474178; PubMed Central PMCID: PMC12142934
8. Lam T, Harmancey R, Vasquez H, Gilbert B, Patel N, Hariharan V. et al. Reversal of intramyocellular lipid accumulation by lipophagy and a p62-mediated pathway. Cell Death Discov. 2016Aug22;2(1):16061 doi:10.1038/cddiscovery.2016.61
9. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011Feb;13(2):132-41 doi:10.1038/ncb2152
10. Maday S, Holzbaur ELF. Compartment-Specific Regulation of Autophagy in Primary Neurons. J Neurosci. 2016Jun1;36(22):5933-45
11. Raimondi M, Cesselli D, Di Loreto C, La Marra F, Schneider C, Demarchi F. USP1 (ubiquitin specific peptidase 1) targets ULK1 and regulates its cellular compartmentalization and autophagy. Autophagy. 2018Oct29;15(4):613-30 doi:10.1080/15548627.2018.1535291 PubMed PMID: 30335599; PubMed Central PMCID: PMC6526860
12. Jaber N, Zong WX. Class III PI3K Vps34: essential roles in autophagy, endocytosis, and heart and liver function. Ann N Y Acad Sci. 2013Mar;1280:48-51 doi:10.1111/nyas.12026 PubMed PMID: 23551104; PubMed Central PMCID: PMC6686116
13. Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011Apr;18(4):571-80 doi:10.1038/cdd.2010.191
14. Jaeger PA, Wyss-Coray T. Beclin 1 Complex in Autophagy and Alzheimer Disease. Arch Neurol. 2010Oct1;67(10):1181-4 doi:10.1001/archneurol.2010.258
15. Iriondo MN, Etxaniz A, Varela YR, Ballesteros U, Lázaro M, Valle M. et al. Effect of ATG12-ATG5-ATG16L1 autophagy E3-like complex on the ability of LC3/GABARAP proteins to induce vesicle tethering and fusion. Cell Mol Life Sci CMLS. 2023Feb2;80(2):56 doi:10.1007/s00018-023-04704-z PubMed PMID: 36729310; PubMed Central PMCID: PMC9894987
16. Yang J, Chen X, Xu H. SQSTM1/p62 droplet -mediated autophagosome formation:insights into Huntington disease. Autophagy. 17(10):3256-9. doi:10.1080/15548627. 2021 1953820 PubMed PMID: 34281469; PubMed Central PMCID: PMC8525954
17. Mailler E, Guardia CM, Bai X, Jarnik M, Williamson CD, Li Y. et al. The autophagy protein ATG9A enables lipid mobilization from lipid droplets. Nat Commun. 2021Nov19;12:6750 doi:10.1038/s41467-021-26999-x PubMed PMID: 34799570; PubMed Central PMCID: PMC8605025
18. Schaaf MBE, Keulers TG, Vooijs MA, Rouschop KMA. LC3/GABARAP family proteins: autophagy-(un)related functions. FASEB J. 2016Dec;30(12):3961-78 doi:10.1096/fj.201600698R
19. Proenca CC, Stoehr N, Bernhard M, Seger S, Genoud C, Roscic A. et al. Atg4b-Dependent Autophagic Flux Alleviates Huntington's Disease Progression. Lim KL, editor. PLoS ONE. 2013Jul8;8(7):e68357 doi:10.1371/journal.pone.0068357
20. Zheng X, Yang Z, Gu Q, Xia F, Fu Y, Liu P. et al. The protease activity of human ATG4B is regulated by reversible oxidative modification. Autophagy. 16(10):1838-50. doi:10.1080/15548627. 2019 1709763 PubMed PMID: 31880198; PubMed Central PMCID: PMC8386634
21. Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Øvervatn A. et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005Nov21;171(4):603-14 doi:10.1083/jcb.200507002
22. Gal J, Strom AL, Kwinter DM, Kilty R, Zhang J, Shi P. et al. Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J Neurochem. 2009Nov;111(4):1062-73 doi:10.1111/j.1471-4159.2009.06388.x PubMed PMID: 19765191; PubMed Central PMCID: PMC2766427
23. Wong YC, Holzbaur ELF. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci. 2014Oct21;111(42):E4439-48 doi:10.1073/pnas.1405752111
24. Thurston TLM, Ryzhakov G, Bloor S, Von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009Nov;10(11):1215-21 doi:10.1038/ni.1800
25. Heo JM, Ordureau A, Swarup S, Paulo JA, Shen K, Sabatini DM. et al. RAB7A phosphorylation by TBK1 promotes mitophagy via the PINK-PARKIN pathway. Sci Adv. 2018Nov2;4(11):eaav0443 doi:10.1126/sciadv.aav0443
26. Su X, Ditlev JA, Hui E, Xing W, Banjade S, Okrut J. et al. Phase separation of signaling molecules promotes T cell receptor signal transduction. Science. 2016Apr29;352(6285):595-9 doi:10.1126/science.aad9964
27. Liu-Yesucevitz L, Lin AY, Ebata A, Boon JY, Reid W, Xu YF. et al. ALS-Linked Mutations Enlarge TDP-43-Enriched Neuronal RNA Granules in the Dendritic Arbor. J Neurosci. 2014Mar19;34(12):4167-74 doi:10.1523/JNEUROSCI.2350-13.2014
28. Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA. et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014Apr28;205(2):143-53 doi:10.1083/jcb.201402104
29. Jiang J, Ballinger CA, Wu Y, Dai Q, Cyr DM, Höhfeld J. et al. CHIP Is a U-box-dependent E3 Ubiquitin Ligase. J Biol Chem. 2001Nov;276(46):42938-44 doi:10.1074/jbc.M101968200
30. Imai Y, Soda M, Hatakeyama S, Akagi T, Hashikawa T, Nakayama KI. et al. CHIP Is Associated with Parkin, a Gene Responsible for Familial Parkinson's Disease, and Enhances Its Ubiquitin Ligase Activity. Mol Cell. 2002Jul1;10(1):55-67 doi:10.1016/S1097-2765(02)00583-X
31. Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL. et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011Mar;471(7340):591-6 doi:10.1038/nature09816
32. Ikeda F, Deribe YL, Skånland SS, Stieglitz B, Grabbe C, Franz-Wachtel M. et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature. 2011Mar;471(7340):637-41 doi:10.1038/nature09814
33. IWAI K. LUBAC-mediated linear ubiquitination: a crucial regulator of immune signaling. Proc Jpn Acad Ser B Phys Biol Sci. 2021Mar11;97(3):120-33 doi:10.2183/pjab.97.007 PubMed PMID: 33692228; PubMed Central PMCID: PMC8019854
34. Li Z, Liu X, Zhu Y, Du Y, Liu X, Lv L. et al. Mitochondrial Phosphoenolpyruvate Carboxykinase Regulates Osteogenic Differentiation by Modulating AMPK/ULK1-Dependent Autophagy. Stem Cells Dayt Ohio. 2019Dec;37(12):1542-55 doi:10.1002/stem.3091 PubMed PMID: 31574189; PubMed Central PMCID: PMC6916635
35. Jiao H, Su GQ, Dong W, Zhang L, Xie W, Yao L m. et al. Chaperone-like protein p32 regulates ULK1 stability and autophagy. Cell Death Differ. 2015Nov;22(11):1812-23 doi:10.1038/cdd.2015.34 PubMed PMID: 25909887; PubMed Central PMCID: PMC4648329
36. Shen H, Xie Y, Wang Y, Xie Y, Wang Y, Su Z. et al. The ER protein CANX (calnexin)-mediated autophagy protects against alzheimer disease. Autophagy. 2025May4;21(5):1096-115 doi:10.1080/15548627.2024.2447206
37. Ryu HY, Kim LE, Jeong H, Yeo BK, Lee JW, Nam H. et al. GSK3B induces autophagy by phosphorylating ULK1. Exp Mol Med. 2021Mar2;53(3):369-83 doi:10.1038/s12276-021-00570-6 PubMed PMID: 33654220; PubMed Central PMCID: PMC8080724
38. Poole LP, Bock-Hughes A, Berardi DE, Macleod KF. ULK1 promotes mitophagy via phosphorylation and stabilization of BNIP3. Sci Rep. 2021Oct15;11:20526 doi:10.1038/s41598-021-00170-4 PubMed PMID: 34654847; PubMed Central PMCID: PMC8519931
39. Li Z, Miao Z, Ding L, Teng X, Bao J. Energy metabolism disorder mediated ammonia gas-induced autophagy via AMPK/mTOR/ULK1-Beclin1 pathway in chicken livers. Ecotoxicol Environ Saf. 2021Jul1;217:112219 doi:10.1016/j.ecoenv.2021.112219
40. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018Mar;25(3):486-541 doi:10.1038/s41418-017-0012-4
41. Takuma K, Yan SS, Stern DM, Yamada K. Mitochondrial Dysfunction, Endoplasmic Reticulum Stress, and Apoptosis in Alzheimer's Disease. J Pharmacol Sci. 2005;97(3):312-6 doi:10.1254/jphs.CPJ04006X
42. Salminen A, Kaarniranta K, Kauppinen A, Ojala J, Haapasalo A, Soininen H. et al. Impaired autophagy and APP processing in Alzheimer's disease: The potential role of Beclin 1 interactome. Prog Neurobiol. 2013Jul1;106-107:33-54 doi:10.1016/j.pneurobio.2013.06.002
43. Leber B, Andrews DW. Closing in on the link between apoptosis and autophagy. F1000 Biol Rep. 2010Dec17;2:88 doi:10.3410/B2-88 PubMed PMID: 21283600; PubMed Central PMCID: PMC3026626
44. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N. et al. Bcl-2 Antiapoptotic Proteins Inhibit Beclin 1-Dependent Autophagy. Cell. 2005Sep23;122(6):927-39 doi:10.1016/j.cell.2005.07.002
45. Malik SA, Shen S, Mariño G, BenYounès A, Maiuri MC, Kroemer G. BH3 mimetics reveal the network properties of autophagy-regulatory signaling cascades. Autophagy. 2011Aug;7(8):914-6 doi:10.4161/auto.7.8.15785
46. Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L. et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006Oct;8(10):1124-32 doi:10.1038/ncb1482
47. Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I. et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010Jan21;1(1):e18-e18 doi:10.1038/cddis.2009.16
48. Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M. et al. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009 10(3)
49. Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ. et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010Feb;12(2):119-31 doi:10.1038/ncb2012
50. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008Dec1;183(5):795-803 doi:10.1083/jcb.200809125
51. Vives-Bauza C, Zhou C, Huang Y, Cui M, De Vries RLA, Kim J. et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci. 2010Jan5;107(1):378-83 doi:10.1073/pnas.0911187107
52. Murakawa T, Yamaguchi O, Hashimoto A, Hikoso S, Takeda T, Oka T. et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun. 2015Jul6;6:7527 doi:10.1038/ncomms8527 PubMed PMID: 26146385; PubMed Central PMCID: PMC4501433
53. Xu L, Pu J. Alpha-Synuclein in Parkinson's Disease: From Pathogenetic Dysfunction to Potential Clinical Application. Park Dis. 2016;2016:1-10 doi:10.1155/2016/1720621
54. Lee VMY, Trojanowski JQ. Mechanisms of Parkinson's Disease Linked to Pathological α-Synuclein: New Targets for Drug Discovery. Neuron. 2006Oct5;52(1):33-8 doi:10.1016/j.neuron.2006.09.026
55. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ. et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017Oct5;171(2):273-85 doi:10.1016/j.cell.2017.09.021
56. Federico G, Carrillo F, Dapporto F, Chiariello M, Santoro M, Bellelli R. et al. NCOA4 links iron bioavailability to DNA metabolism. Cell Rep. 2022Aug16;40(7):111207 doi:10.1016/j.celrep.2022.111207
57. Li X, Chu Y, Ma R, Dou M, Li S, Song Y. et al. Ferroptosis as a mechanism of oligodendrocyte loss and demyelination in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2022Dec15;373:577995 doi:10.1016/j.jneuroim.2022.577995
58. Zille M, Kumar A, Kundu N, Bourassa MW, Wong VSC, Willis D. et al. Ferroptosis in Neurons and Cancer Cells Is Similar But Differentially Regulated by Histone Deacetylase Inhibitors. eNeuro. 2019Feb15;6(1):ENEURO.0263-18.2019 doi:10.1523/ENEURO.0263-18.2019 PubMed PMID: 30783618; PubMed Central PMCID: PMC6378329
59. Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K. et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002Jun1;16(11):1345-55 doi:10.1101/gad.992302 PubMed PMID: 12050113; PubMed Central PMCID: PMC186318
60. Andrabi SA, Umanah GKE, Chang C, Stevens DA, Karuppagounder SS, Gagné JP. et al. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci. 2014Jul15;111(28):10209-14 doi:10.1073/pnas.1405158111
61. Wu MY, Song JX, Wang SF, Cai CZ, Li M, Lu JH. Selective autophagy: The new player in the fight against neurodegenerative diseases? Brain Res Bull. 2018Mar;137:79-90 doi:10.1016/j.brainresbull.2017.11.009
62. Kim MJ, Yoon JH, Ryu JH. Mitophagy: a balance regulator of NLRP3 inflammasome activation. BMB Rep. 2016Oct31;49(10):529-35 doi:10.5483/BMBRep.2016.49.10.115 PubMed PMID: 27439607; PubMed Central PMCID: PMC5227293
63. Haque MdE, Akther M, Jakaria Md, Kim IS, Azam S, Choi DK. Targeting the Microglial NLRP3 Inflammasome and Its Role in Parkinson's Disease. Mov Disord. 2020;35(1):20-33 doi:10.1002/mds.27874
64. Tao G, Wang X, Wang J, Ye Y, Zhang M, Lang Y. et al. Dihydro-resveratrol ameliorates NLRP3 inflammasome-mediated neuroinflammation via Bnip3-dependent mitophagy in Alzheimer's disease. Br J Pharmacol. 2025Feb;182(4):1005-24 doi:10.1111/bph.17373
65. Bright F, Chan G, Van Hummel A, Ittner LM, Ke YD. TDP-43 and Inflammation: Implications for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Int J Mol Sci. 2021Jul21;22(15):7781 doi:10.3390/ijms22157781
66. Xu W, Ocak U, Gao L, Tu S, Lenahan CJ, Zhang J. et al. Selective autophagy as a therapeutic target for neurological diseases. Cell Mol Life Sci CMLS. 2020Oct16;78(4):1369-92 doi:10.1007/s00018-020-03667-9 PubMed PMID: 33067655; PubMed Central PMCID: PMC7904548
67. Hergesheimer RC, Chami AA, de Assis DR, Vourc'h P, Andres CR, Corcia P. et al. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: a resolution in sight? Brain. 2019May;142(5):1176-94 doi:10.1093/brain/awz078 PubMed PMID: 30938443; PubMed Central PMCID: PMC6487324
68. Zhou X, Wang J, Yu L, Qiao G, Qin D, Yuen-Kwan Law B. et al. Mitophagy and cGAS-STING crosstalk in neuroinflammation. Acta Pharm Sin B. 2024Aug;14(8):3327-61 doi:10.1016/j.apsb.2024.05.012 PubMed PMID: 39220869; PubMed Central PMCID: PMC11365416
69. Woo MS, Mayer C, Binkle-Ladisch L, Sonner JK, Rosenkranz SC, Shaposhnykov A. et al. STING orchestrates the neuronal inflammatory stress response in multiple sclerosis. Cell. 2024Jul;187(15):4043-4060.e30 doi:10.1016/j.cell.2024.05.031
70. Wang R, Sun H, Cao Y, Zhang Z, Chen Y, Wang X. et al. Glucosylceramide accumulation in microglia triggers STING-dependent neuroinflammation and neurodegeneration in mice. Sci Signal. 2024Mar26;17(829):eadk8249 doi:10.1126/scisignal.adk8249
71. Jin M meng, Wang F, Qi D, Liu W wen, Gu C, Mao CJ. et al. A Critical Role of Autophagy in Regulating Microglia Polarization in Neurodegeneration. Front Aging Neurosci. 2018Nov20;10:378 doi:10.3389/fnagi.2018.00378
72. Zubova SG, Suvorova II, Karpenko MN. Macrophage and microglia polarization: focus on autophagy-dependent reprogramming. IMR Press. 2022Jan20;14(1):3 doi:10.31083/j.fbs1401003
73. Li J, O W, Li W, Jiang ZG, Ghanbari HA. Oxidative Stress and Neurodegenerative Disorders. Int J Mol Sci. 2013Dec16;14(12):24438-75 doi:10.3390/ijms141224438
74. Wang XL, Feng ST, Wang YT, Yuan YH, Li ZP, Chen NH. et al. Mitophagy, a Form of Selective Autophagy, Plays an Essential Role in Mitochondrial Dynamics of Parkinson's Disease. Cell Mol Neurobiol. 2021Feb2;42(5):1321-39 doi:10.1007/s10571-021-01039-w PubMed PMID: 33528716; PubMed Central PMCID: PMC11421754
75. Jiang T, Harder B, Rojo de la Vega M, Wong PK, Chapman E, Zhang DD. p62 links autophagy and Nrf2 signaling. Free Radic Biol Med. 2015Nov;88(0 0):199-204 doi:10.1016/j.freeradbiomed.2015.06.014 PubMed PMID: 26117325; PubMed Central PMCID: PMC4628872
76. Shah SZA, Zhao D, Hussain T, Sabir N, Mangi MH, Yang L. p62-Keap1-NRF2-ARE Pathway: A Contentious Player for Selective Targeting of Autophagy, Oxidative Stress and Mitochondrial Dysfunction in Prion Diseases. Front Mol Neurosci. 2018Oct4;11:310 doi:10.3389/fnmol.2018.00310
77. Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci. 2016Sep;73(17):3221-47 doi:10.1007/s00018-016-2223-0
78. Guo F, Liu X, Cai H, Le W. Autophagy in neurodegenerative diseases: pathogenesis and therapy. Brain Pathol. 2018Jan;28(1):3-13 doi:10.1111/bpa.12545
79. Cuervo AM. Autophagy and aging: keeping that old broom working. Trends Genet. 2008Dec1;24(12):604-12 doi:10.1016/j.tig.2008.10.002
80. Supruniuk E, Baczewska M, Żebrowska E, Maciejczyk M, Lauko KK, Dajnowicz-Brzezik P. et al. Redox Biomarkers and Matrix Remodeling Molecules in Ovarian Cancer. Antioxidants. 2024Feb4;13(2):200 doi:10.3390/antiox13020200 PubMed PMID: 38397798; PubMed Central PMCID: PMC10885995
81. Guo C, Sun L, Chen X, Zhang D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res. 2013Jul25;8(21):2003-14 doi:10.3969/j.issn.1673-5374.2013.21.009 PubMed PMID: 25206509; PubMed Central PMCID: PMC4145906
82. Edens BM, Miller N, Ma YC. Impaired Autophagy and Defective Mitochondrial Function: Converging Paths on the Road to Motor Neuron Degeneration. Front Cell Neurosci. 2016Mar3;10:44 doi:10.3389/fncel.2016.00044
83. Genin EC, Abou-Ali M, Paquis-Flucklinger V. Mitochondria, a Key Target in Amyotrophic Lateral Sclerosis Pathogenesis. Genes. 2023Oct24;14(11):1981 doi:10.3390/genes14111981 PubMed PMID: 38002924; PubMed Central PMCID: PMC10671245
84. Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007Apr4;26(7):1749-60 doi:10.1038/sj.emboj.7601623
85. He J, Liu J, Huang Y, Tang X, Xiao H, Hu Z. Oxidative Stress, Inflammation, and Autophagy: Potential Targets of Mesenchymal Stem Cells-Based Therapies in Ischemic Stroke. Front Neurosci. 2021Feb26;15:641157 doi:10.3389/fnins.2021.641157
86. Seco-Cervera M, González-Cabo P, Pallardó FV, Romá-Mateo C, García-Giménez JL. Thioredoxin and Glutaredoxin Systems as Potential Targets for the Development of New Treatments in Friedreich's Ataxia. Antioxidants. 2020Dec10;9(12):1257 doi:10.3390/antiox9121257 PubMed PMID: 33321938; PubMed Central PMCID: PMC7763308
87. Bjørklund G, Zou L, Peana M, Chasapis CT, Hangan T, Lu J. et al. The Role of the Thioredoxin System in Brain Diseases. Antioxidants. 2022Oct31;11(11):2161 doi:10.3390/antiox11112161 PubMed PMID: 36358532; PubMed Central PMCID: PMC9686621
88. Dafre AL, Schmitz AE, Maher P. Methylglyoxal-induced AMPK activation leads to autophagic degradation of thioredoxin 1 and glyoxalase 2 in HT22 nerve cells. Free Radic Biol Med. 2017Jul;108:270-9 doi:10.1016/j.freeradbiomed.2017.03.028 PubMed PMID: 28363601; PubMed Central PMCID: PMC5492945
89. Cunha-Oliveira T, Montezinho L, Simões RF, Carvalho M, Ferreiro E, Silva FSG. Mitochondria: A Promising Convergent Target for the Treatment of Amyotrophic Lateral Sclerosis. Cells. 2024Jan29;13(3):248 doi:10.3390/cells13030248
90. Cenini G, Lloret A, Cascella R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxid Med Cell Longev. 2019May9;2019:1-18 doi:10.1155/2019/2105607
91. Wu Q, Gao C, Wang H, Zhang X, Li Q, Gu Z. et al. Mdivi-1 alleviates blood-brain barrier disruption and cell death in experimental traumatic brain injury by mitigating autophagy dysfunction and mitophagy activation. Int J Biochem Cell Biol. 2018Jan1;94:44-55 doi:10.1016/j.biocel.2017.11.007
92. Yang Z, Lin P, Chen B, Zhang X, Xiao W, Wu S. et al. Autophagy alleviates hypoxia-induced blood-brain barrier injury via regulation of CLDN5 (claudin 5). Autophagy. 17(10):3048-67. doi:10.1080/15548627. 2020 1851897 PubMed PMID: 33280500; PubMed Central PMCID: PMC8526012
93. Kim KA, Shin D, Kim JH, Shin YJ, Rajanikant GK, Majid A. et al. Role of Autophagy in Endothelial Damage and Blood-Brain Barrier Disruption in Ischemic Stroke. Stroke. 2018Jun;49(6):1571-9 doi:10.1161/STROKEAHA.117.017287
94. Chen CY, Chao YM, Lin HF, Chen CJ, Chen CS, Yang JL. et al. miR-195 reduces age-related blood-brain barrier leakage caused by thrombospondin-1-mediated selective autophagy. Aging Cell. 2020;19(11):e13236 doi:10.1111/acel.13236
95. Van Hameren G, Aboghazleh R, Parker E, Dreier JP, Kaufer D, Friedman A. From spreading depolarization to blood-brain barrier dysfunction: navigating traumatic brain injury for novel diagnosis and therapy. Nat Rev Neurol. 2024Jul;20(7):408-25 doi:10.1038/s41582-024-00973-9
96. Kim J, Kim HS, Chung JH. Molecular mechanisms of mitochondrial DNA release and activation of the cGAS-STING pathway. Exp Mol Med. 2023Mar24;55(3):510-9 doi:10.1038/s12276-023-00965-7 PubMed PMID: 36964253; PubMed Central PMCID: PMC10037406
97. Clark EH, Vázquez de la Torre A, Hoshikawa T, Briston T. Targeting mitophagy in Parkinson's disease. J Biol Chem. 2020Dec24;296:100209 doi:10.1074/jbc.REV120.014294 PubMed PMID: 33372898; PubMed Central PMCID: PMC7948953
98. Wasner K, Smajic S, Ghelfi J, Delcambre S, Prada-Medina CA, Knappe E. et al. Parkin Deficiency Impairs Mitochondrial DNA Dynamics and Propagates Inflammation. Mov Disord. 2022Jul;37(7):1405-15 doi:10.1002/mds.29025
99. Wang S, Long H, Hou L, Feng B, Ma Z, Wu Y. et al. The mitophagy pathway and its implications in human diseases. Signal Transduct Target Ther. 2023Aug16;8:304 doi:10.1038/s41392-023-01503-7 PubMed PMID: 37582956; PubMed Central PMCID: PMC10427715
100. Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK. et al. Parkin Protein Deficiency Exacerbates Cardiac Injury and Reduces Survival following Myocardial Infarction. J Biol Chem. 2013Jan11;288(2):915-26 doi:10.1074/jbc.M112.411363 PubMed PMID: 23152496; PubMed Central PMCID: PMC3543040
101. Chen Z, Li G. Immune response and blood-brain barrier dysfunction during viral neuroinvasion. Innate Immun. 2021Feb;27(2):109-17 doi:10.1177/1753425920954281 PubMed PMID: 32903111; PubMed Central PMCID: PMC7882805
102. Wakida NM, Lau AL, Nguyen J, Cruz GMS, Fote GM, Steffan JS. et al. Diminished LC3-Associated Phagocytosis by Huntington's Disease Striatal Astrocytes. J Huntingt Dis. 11(1):25-33. doi:10.3233/JHD-210502 PubMed PMID: 35253772; PubMed Central PMCID: PMC9028675.
103. Szabó Á, Vincze V, Chhatre AS, Jipa A, Bognár S, Varga KE. et al. LC3-associated phagocytosis promotes glial degradation of axon debris after injury in Drosophila models. Nat Commun. 2023May29;14:3077 doi:10.1038/s41467-023-38755-4 PubMed PMID: 37248218; PubMed Central PMCID: PMC10227080
104. Bertrand L, Dygert L, Toborek M. Antiretroviral Treatment with Efavirenz Disrupts the Blood-Brain Barrier Integrity and Increases Stroke Severity. Sci Rep. 2016Dec23;6(1):39738 doi:10.1038/srep39738
105. Shusta EV. Blood-brain barrier genomics, proteomics, and new transporter discovery. Vol. 2. 2005 2(1)
106. Zou L, Liao M, Zhen Y, Zhu S, Chen X, Zhang J. et al. Autophagy and beyond: Unraveling the complexity of UNC-51-like kinase 1 (ULK1) from biological functions to therapeutic implications. Acta Pharm Sin B. 2022Oct;12(10):3743-82 doi:10.1016/j.apsb.2022.06.004 PubMed PMID: 36213540; PubMed Central PMCID: PMC9532564
107. Lu W, Chu H, Yang C, Li X. Transcription factor EB (TFEB) promotes autophagy in early brain injury after subarachnoid hemorrhage in rats. Neurosurg Rev. 2024
108. Hwang JY, Yan J, Zukin RS. Autophagy and synaptic plasticity: epigenetic regulation. Curr Opin Neurobiol. 2019Dec;59:207-12 doi:10.1016/j.conb.2019.09.010 PubMed PMID: 31634675; PubMed Central PMCID: PMC7597971
109. Sun F, Huo X, Zhai Y, Wang A, Xu J, Su D. et al. Crystal Structure of Mitochondrial Respiratory Membrane Protein Complex II. Cell. 2005Jul1;121(7):1043-57 doi:10.1016/j.cell.2005.05.025
110. Nikoletopoulou V, Sidiropoulou K, Kallergi E, Dalezios Y, Tavernarakis N. Modulation of Autophagy by BDNF Underlies Synaptic Plasticity. Cell Metab. 2017Jul5;26(1):230-242.e5 doi:10.1016/j.cmet.2017.06.005
111. Meraz-Ríos MA, Lira-De León KI, Campos-Peña V, De Anda-Hernández MA, Mena-López R. Tau oligomers and aggregation in Alzheimer's disease. J Neurochem. 2010Mar;112(6):1353-67 doi:10.1111/j.1471-4159.2009.06511.x
112. Guerrero-Muñoz MJ, Gerson J, Castillo-Carranza DL. Tau Oligomers: The Toxic Player at Synapses in Alzheimer's Disease. Front Cell Neurosci. 2015Dec2;9:464 doi:10.3389/fncel.2015.00464
113. Xu P, Zhang T, Yu F, Guo L, Yang Y. ATG9 promotes autophagosome formation through interaction with LC3. Biochem Biophys Res Commun. 2025Feb2;747:151254 doi:10.1016/j.bbrc.2024.151254
114. Rowlands J, Moore DJ. VPS35 and retromer dysfunction in Parkinson's disease. Philos Trans R Soc B Biol Sci. 2024Apr8;379(1899):20220384 doi:10.1098/rstb.2022.0384
115. Chen BS, Thomas EV, Sanz-Clemente A, Roche KW. NMDA Receptor-Dependent Regulation of Dendritic Spine Morphology by SAP102 Splice Variants. J Neurosci. 2011Jan5;31(1):89-96 doi:10.1523/JNEUROSCI.1034-10.2011 PubMed PMID: 21209193; PubMed Central PMCID: PMC3030119
116. Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N. et al. Arc/Arg3.1 Interacts with the Endocytic Machinery to Regulate AMPA Receptor Trafficking. Neuron. 2006Nov9;52(3):445-59 doi:10.1016/j.neuron.2006.08.033
117. Wall MJ, Corrêa SAL. The mechanistic link between Arc/Arg3.1 expression and AMPA receptor endocytosis. Semin Cell Dev Biol. 2018 May 1;Arc/ARg3.177:17-24. doi:10.1016/j.semcdb.2017.09.005
118. Hou SJ, Zhang SX, Li Y, Xu SY. Rapamycin Responds to Alzheimer's Disease: A Potential Translational Therapy. Clin Interv Aging. 2023Oct2;18:1629-39 doi:10.2147/CIA.S429440 PubMed PMID: 37810956; PubMed Central PMCID: PMC10557994
119. Wang X, Xia W, Li K, Zhang Y, Ge W, Ma C. Rapamycin regulates cholesterol biosynthesis and cytoplasmic ribosomal proteins in hippocampus and temporal lobe of APP/PS1 mouse. J Neurol Sci. 2019Apr15;399:125-39 doi:10.1016/j.jns.2019.02.022
120. Li J, Zhang D, Wiersma M, Brundel BJJM. Role of Autophagy in Proteostasis: Friend and Foe in Cardiac Diseases. Cells. 2018Dec19;7(12):279 doi:10.3390/cells7120279 PubMed PMID: 30572675; PubMed Central PMCID: PMC6316637
121. Panwar S, Uniyal P, Kukreti N, Hashmi A, Verma S, Arya A. et al. Role of autophagy and proteostasis in neurodegenerative diseases: Exploring the therapeutic interventions. Chem Biol Drug Des. 2024;103(4):e14515 doi:10.1111/cbdd.14515
122. Kumar AV, Mills J, Lapierre LR. Selective Autophagy Receptor p62/SQSTM1, a Pivotal Player in Stress and Aging. Front Cell Dev Biol. 2022Feb14;10:793328 doi:10.3389/fcell.2022.793328
123. Jeong SJ, Zhang X, Rodriguez-Velez A, Evans TD, Razani B. p62/SQSTM1 and Selective Autophagy in Cardiometabolic Diseases. Antioxid Redox Signal. 2019Aug20;31(6):458-71 doi:10.1089/ars.2018.7649 PubMed PMID: 30588824; PubMed Central PMCID: PMC6653798
124. Faruk MO, Ichimura Y, Kageyama S, Komatsu-Hirota S, El-Gowily AH, Sou Y shin. et al. Phase-separated protein droplets of amyotrophic lateral sclerosis-associated p62/SQSTM1 mutants show reduced inner fluidity. J Biol Chem. 2021Dec1;297(6):101405 doi:10.1016/j.jbc.2021.101405
125. Deng Z, Lim J, Wang Q, Purtell K, Wu S, Palomo GM. et al. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy. 2019Jul30;16(5):917-31 doi:10.1080/15548627.2019.1644076 PubMed PMID: 31362587; PubMed Central PMCID: PMC7144840
126. Assaye MA, Gizaw ST. Chaperone-Mediated Autophagy and Its Implications for Neurodegeneration and Cancer. Int J Gen Med. 2022Jun15;15:5635-49 doi:10.2147/IJGM.S368364 PubMed PMID: 35734200; PubMed Central PMCID: PMC9207255
127. Dompierre JP, Godin JD, Charrin BC, Cordelières FP, King SJ, Humbert S. et al. Histone Deacetylase 6 Inhibition Compensates for the Transport Deficit in Huntington's Disease by Increasing Tubulin Acetylation. J Neurosci. 2007Mar28;27(13):3571-83
128. Simões-Pires C, Zwick V, Nurisso A, Schenker E, Carrupt PA, Cuendet M. HDAC6 as a target for neurodegenerative diseases: what makes it different from the other HDACs? Mol Neurodegener. 2013Jan29;8(1):7 doi:10.1186/1750-1326-8-7
129. Li Z, Wang C, Wang Z, Zhu C, Li J, Sha T. et al. Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds. Nature. 2019Nov7;575(7781):203-9 doi:10.1038/s41586-019-1722-1
130. Maday S, Holzbaur ELF. Autophagosome assembly and cargo capture in the distal axon. Autophagy. 2012May1;8(5):858-60 doi:10.4161/auto.20055 PubMed PMID: 22617438; PubMed Central PMCID: PMC3378425
131. Stavoe AKH, Holzbaur ELF. Neuronal autophagy declines substantially with age and is rescued by overexpression of WIPI2. Autophagy. 2019Dec3;16(2):371-2 doi:10.1080/15548627.2019.1695401 PubMed PMID: 31794336; PubMed Central PMCID: PMC6984449
132. Palmer JE, Wilson N, Son SM, Obrocki P, Wrobel L, Rob M. et al. Autophagy, aging, and age-related neurodegeneration. Neuron. 2025Jan;113(1):29-48 doi:10.1016/j.neuron.2024.09.015
133. Heckmann BL, Teubner BJW, Tummers B, Boada-Romero E, Harris L, Yang M. et al. LC3-associated endocytosis facilitates β-amyloid clearance and mitigates neurodegeneration in murine Alzheimer's Disease. Cell. 2019Jul25;178(3):536-551.e14 doi:10.1016/j.cell.2019.05.056 PubMed PMID: 31257024; PubMed Central PMCID: PMC6689199
134. Plaza-Zabala A, Sierra-Torre V, Sierra A. Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging. Int J Mol Sci. 2017Mar9;18(3):598 doi:10.3390/ijms18030598 PubMed PMID: 28282924; PubMed Central PMCID: PMC5372614
135. Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T. et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015Nov;18(11):1584-93 doi:10.1038/nn.4132 PubMed PMID: 26436904; PubMed Central PMCID: PMC4694577
136. Qian J, Ren S, Ren T, Shi R, Qiao L, Kang J. Astrocyte Autophagy in Neurodegenerative Diseases: Current Progress in Mechanisms and Therapeutics. Neurochem Res. 2025Oct;50(5):287 doi:10.1007/s11064-025-04532-6
137. Guglielmotto M, Monteleone D, Piras A, Valsecchi V, Tropiano M, Ariano S. et al. Aβ1-42 monomers or oligomers have different effects on autophagy and apoptosis. Autophagy. 2014Oct1;10(10):1827-43 doi:10.4161/auto.30001 PubMed PMID: 25136804; PubMed Central PMCID: PMC4198366
138. Madadi S, Schwarzenbach H, Saidijam M, Mahjub R, Soleimani M. Potential microRNA-related targets in clearance pathways of amyloid-β: novel therapeutic approach for the treatment of Alzheimer's disease. Cell Biosci. 2019Nov12;9:91 doi:10.1186/s13578-019-0354-3 PubMed PMID: 31749959; PubMed Central PMCID: PMC6852943
139. Huang Z, Ang W, Huang H, Wang Y. VD/VDR-mediated ATG16L1 activation reduces Alzheimer's disease-like pathology and cognitive decline. Mol Cell Toxicol. 2024Feb24;21(1):151-62 doi:10.1007/s13273-024-00429-7
140. Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM. et al. Lysosomal Proteolysis and Autophagy Require Presenilin 1 and Are Disrupted by Alzheimer-Related PS1 Mutations. Cell. 2010Jun25;141(7):1146-58 doi:10.1016/j.cell.2010.05.008 PubMed PMID: 20541250; PubMed Central PMCID: PMC3647462
141. Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired Degradation of Mutant α-Synuclein by Chaperone-Mediated Autophagy. Science. 2004Aug27;305:1292-5 Located at: world. doi:10.1126/science.1101738
142. Heneka MT, McManus RM, Latz E. Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci. 2018Oct;19(10):610-21 doi:10.1038/s41583-018-0055-7
143. Song M, Zhao X, Song F. Aging-Dependent Mitophagy Dysfunction in Alzheimer's Disease. Mol Neurobiol. 2021May;58(5):2362-78 doi:10.1007/s12035-020-02248-y
144. Lautrup S, Lou G, Aman Y, Nilsen H, Tao J, Fang EF. Microglial mitophagy mitigates neuroinflammation in Alzheimer's disease. Neurochem Int. 2019Oct1;129:104469 doi:10.1016/j.neuint.2019.104469
145. Fang EF, Lautrup S, Hou Y, Demarest TG, Croteau DL, Mattson MP. et al. NAD+ in Aging: Molecular Mechanisms and Translational Implications. Trends Mol Med. 2017Oct1;23(10):899-916 doi:10.1016/j.molmed.2017.08.001
146. Hou X, Watzlawik JO, Fiesel FC, Springer W. Autophagy in Parkinson's Disease. J Mol Biol. 2020 Apr 3;Autophagy in Neurodegenerative Diseases432(8):2651-72. doi:10.1016/j. jmb. 2020 01.037
147. Sakurai M, Kuwahara T. Canonical and noncanonical autophagy: involvement in Parkinson's disease. Front Cell Dev Biol. 2025Jan30;13:1518991 doi:10.3389/fcell.2025.1518991
148. Teravskis PJ, Covelo A, Miller EC, Singh B, Martell-Martínez HA, Benneyworth MA. et al. A53T Mutant Alpha-Synuclein Induces Tau-Dependent Postsynaptic Impairment Independently of Neurodegenerative Changes. J Neurosci. 2018Nov7;38(45):9754-67 doi:10.1523/JNEUROSCI.0344-18.2018 PubMed PMID: 30249789; PubMed Central PMCID: PMC6222065
149. Choi I, Heaton GR, Lee YK, Yue Z. Regulation of α-synuclein homeostasis and inflammasome activation by microglial autophagy. Sci Adv. 2022Oct26;8(43):eabn1298 doi:10.1126/sciadv.abn1298
150. Kalia LV, Kalia SK. α-Synuclein and Lewy pathology in Parkinson's disease. Curr Opin Neurol. 2015Aug;28(4):375-81 doi:10.1097/WCO.0000000000000215
151. Chen K, Martens YA, Meneses A, Ryu DH, Lu W, Raulin AC. et al. LRP1 is a neuronal receptor for α-synuclein uptake and spread. Mol Neurodegener. 2022Sep2;17:57 doi:10.1186/s13024-022-00560-w PubMed PMID: 36056345; PubMed Central PMCID: PMC9438229
152. Yang K, Yan Y, Yu A, Zhang R, Zhang Y, Qiu Z. et al. Mitophagy in neurodegenerative disease pathogenesis. Neural Regen Res. 2023Sep22;19(5):998-1005 doi:10.4103/1673-5374.385281 PubMed PMID: 37862201; PubMed Central PMCID: PMC10749592
153. El Manaa W, Duplan E, Goiran T, Lauritzen I, Vaillant Beuchot L, Lacas-Gervais S. et al. Transcription- and phosphorylation-dependent control of a functional interplay between XBP1s and PINK1 governs mitophagy and potentially impacts Parkinson disease pathophysiology. Autophagy. 17(12):4363-85. doi:10.1080/15548627. 2021 1917129 PubMed PMID: 34030589; PubMed Central PMCID: PMC8726674
154. Tain LS, Chowdhury RB, Tao RN, Plun-Favreau H, Moisoi N, Martins LM. et al. Drosophila HtrA2 is dispensable for apoptosis but acts downstream of PINK1 independently from Parkin. Cell Death Differ. 2009Aug;16(8):1118-25 doi:10.1038/cdd.2009.23 PubMed PMID: 19282869; PubMed Central PMCID: PMC2711053
155. Li W, Jiang WS, Su YR, Tu KW, Zou L, Liao CR. et al. PINK1/Parkin-mediated mitophagy inhibits osteoblast apoptosis induced by advanced oxidation protein products. Cell Death Dis. 2023Feb7;14(2):88 doi:10.1038/s41419-023-05595-5 PubMed PMID: 36750550; PubMed Central PMCID: PMC9905061
156. Qiao Y, Kou J, Tian Y, Ma W, Yu Y, Pang J. et al. Subcellular localization and function analysis of PINK1 mitron in PD progression: Mitron modulates mitochondrial morphology to regulate neuronal death. J Biol Chem. 2024Oct1;300(10):107773 doi:10.1016/j.jbc.2024.107773
157. Lei L, Yuan J, Dai Z, Xiang S, Tu Q, Cui X. et al. Targeting the Labile Iron Pool with Engineered DFO Nanosheets to Inhibit Ferroptosis for Parkinson's Disease Therapy. Adv Mater. 2024Oct;36(41):2409329 doi:10.1002/adma.202409329
158. Baer AS, Syed YA, Kang SU, Mitteregger D, Vig R, ffrench-Constant C. et al. Myelin-mediated inhibition of oligodendrocyte precursor differentiation can be overcome by pharmacological modulation of Fyn-RhoA and protein kinase C signalling. Brain. 2009Feb1;132(2):465-81 doi:10.1093/brain/awn334
159. López-Muguruza E, Matute C. Alterations of Oligodendrocyte and Myelin Energy Metabolism in Multiple Sclerosis. Int J Mol Sci. 2023Aug18;24(16):12912 doi:10.3390/ijms241612912 PubMed PMID: 37629092; PubMed Central PMCID: PMC10454078
160. Li Q, Zhao Y, Guo H, Li Q, Yan C, Li Y. et al. Impaired lipophagy induced-microglial lipid droplets accumulation contributes to the buildup of TREM1 in diabetes-associated cognitive impairment. Autophagy. 19(10):2639-56. doi:10.1080/15548627. 2023 2213984 PubMed PMID: 37204119; PubMed Central PMCID: PMC10472854
161. Xiao S, Peng K, Li C, Long Y, Yu Q. The role of sphingosine-1-phosphate in autophagy and related disorders. Cell Death Discov. 2023Oct18;9:380 doi:10.1038/s41420-023-01681-x PubMed PMID: 37852968; PubMed Central PMCID: PMC10584985
162. Chen Y, Sun L, Liu H, Li J, Guo L, Wang Z. KLF4 interacts with TXNIP to modulate the pyroptosis in ulcerative colitis via regulating NLRP3 signaling. Immun Inflamm Dis. 2024;12(2):e1199 doi:10.1002/iid3.1199
163. Liang Y, Zhao J, Dai T, Li X, Chen L, He Z. et al. A review of KLF4 and inflammatory disease: Current status and future perspective. Pharmacol Res. 2024Sep1;207:107345 doi:10.1016/j.phrs.2024.107345
164. Wei Y, Zhao X, Zhang Y, Cui C, Han S, Yang C. et al. miR-7 promotes apoptosis and autophagy of granulosa cells by targeting KLF4 via JAK/STAT3 signaling pathway in chickens. Theriogenology. 2024Dec1;230:322-9 doi:10.1016/j.theriogenology.2024.09.032
165. Pandey R, Bakay M, Hakonarson H. CLEC16A-An Emerging Master Regulator of Autoimmunity and Neurodegeneration. Int J Mol Sci. 2023May4;24(9):8224 doi:10.3390/ijms24098224 PubMed PMID: 37175930; PubMed Central PMCID: PMC10179542
166. Yang M, Wei X, Yi X, Jiang DS. Mitophagy-related regulated cell death: molecular mechanisms and disease implications. Cell Death Dis. 2024Jul16;15(7):505 doi:10.1038/s41419-024-06804-5
167. Gingerich MA, Liu X, Chai B, Pearson GL, Vincent MP, Stromer T. et al. An intrinsically disordered protein region encoded by the human disease gene CLEC16A regulates mitophagy. Autophagy. 19(2):525-43. doi:10.1080/15548627. 2022 2080383 PubMed PMID: 35604110; PubMed Central PMCID: PMC9851259
168. Kim KA, Kim D, Kim JH, Shin YJ, Kim ES, Akram M. et al. Autophagy-mediated occludin degradation contributes to blood-brain barrier disruption during ischemia in bEnd.3 brain endothelial cells and rat ischemic stroke models. Fluids Barriers CNS. 2020Mar14;17:21 doi:10.1186/s12987-020-00182-8 PubMed PMID: 32169114; PubMed Central PMCID: PMC7071658
169. Al-kuraishy HM, Jabir MS, Al-Gareeb AI, Saad HM, Batiha GES, Klionsky DJ. The beneficial role of autophagy in multiple sclerosis: Yes or No? Autophagy. 20(2):259-74. doi:10.1080/15548627. 2023 2259281 PubMed PMID: 37712858; PubMed Central PMCID: PMC10813579
170. Peña-Martinez C, Rickman AD, Heckmann BL. Beyond autophagy: LC3-associated phagocytosis and endocytosis. Sci Adv. 8(43):eabn1702. doi:10.1126/sciadv.abn1702 PubMed PMID: 36288309; PubMed Central PMCID: PMC9604515.
171. Chen M, Zhang H, Chu YH, Tang Y, Pang XW, Qin C. et al. Microglial autophagy in cerebrovascular diseases. Front Aging Neurosci. 2022Oct6;14:1023679 doi:10.3389/fnagi.2022.1023679
172. Scotter EL, Chen HJ, Shaw CE. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics. 2015Apr1;12(2):352-63 doi:10.1007/s13311-015-0338-x
173. Ko VI, Ong K, Cleveland DW, Yu H, Ravits JM. CK1δ/ε kinases regulate TDP-43 phosphorylation and are therapeutic targets for ALS-related TDP-43 hyperphosphorylation. Neurobiol Dis. 2024Jun15;196:106516 doi:10.1016/j.nbd.2024.106516
174. Inukai Y, Nonaka T, Arai T, Yoshida M, Hashizume Y, Beach TG. et al. Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett. 2008;582(19):2899-904 doi:10.1016/j.febslet.2008.07.027
175. Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM, Ferraiuolo L. et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016Aug;35(15):1656-76 doi:10.15252/embj.201694401
176. Rashid HO, Yadav RK, Kim HR, Chae HJ. ER stress: Autophagy induction, inhibition and selection. Autophagy. 2015Nov2;11(11):1956-77 doi:10.1080/15548627.2015.1091141 PubMed PMID: 26389781
177. Nishitoh H, Kadowaki H, Nagai A, Maruyama T, Yokota T, Fukutomi H. et al. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008Jun1;22(11):1451-64 doi:10.1101/gad.1640108 PubMed PMID: 18519638; PubMed Central PMCID: PMC2418582
178. Relic B, Deroyer C, Malaise O, Plener Z, Gillet P, de Seny D. et al. TFEB phosphorylation on Serine 211 is induced by autophagy in human synovial fibroblasts and by p62/SQSTM1 overexpression in HEK293 cells. Biochem J. 2021Aug27;478(16):3145-55 doi:10.1042/BCJ20210174 PubMed PMID: 34405859; PubMed Central PMCID: PMC8421036
179. Settembre C, Di Malta C, Polito VA, Arencibia MG, Vetrini F, Erdin S. et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science. 2011Jun17;332(6036):1429-33 doi:10.1126/science.1204592
180. Rusmini P, Cortese K, Crippa V, Cristofani R, Cicardi ME, Ferrari V. et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy. 2019Apr3;15(4):631-51 doi:10.1080/15548627.2018.1535292
181. Yuan Y, Zheng Y, Zhang X, Chen Y, Wu X, Wu J. et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy. 2017Aug18;13(10):1754-66 doi:10.1080/15548627.2017.1357792 PubMed PMID: 28820284; PubMed Central PMCID: PMC5640199
182. Guan R, Zou W, Dai X, Yu X, Liu H, Chen Q. et al. Mitophagy, a potential therapeutic target for stroke. J Biomed Sci. 2018Nov30;25:87 doi:10.1186/s12929-018-0487-4 PubMed PMID: 30501621; PubMed Central PMCID: PMC6271612
183. Buhlman L, Damiano M, Bertolin G, Ferrando-Miguel R, Lombès A, Brice A. et al. Functional interplay between Parkin and Drp1 in mitochondrial fission and clearance. Biochim Biophys Acta BBA - Mol Cell Res. 2014Sep1;1843(9):2012-26 doi:10.1016/j.bbamcr.2014.05.012
184. Hadley G, Beard DJ, Couch Y, Neuhaus AA, Adriaanse BA, DeLuca GC. et al. Rapamycin in ischemic stroke: Old drug, new tricks? J Cereb Blood Flow Metab. 2019Jan;39(1):20-35 doi:10.1177/0271678X18807309 PubMed PMID: 30334673; PubMed Central PMCID: PMC6311672
185. Lin M miao, Liu N, Qin Z hong, Wang Y. Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol Sin. 2022Oct;43(10):2439-47 doi:10.1038/s41401-022-00879-6 PubMed PMID: 35233090; PubMed Central PMCID: PMC9525705
186. Xiong R, Zhou XG, Tang Y, Wu JM, Sun YS, Teng JF. et al. Lychee seed polyphenol protects the blood-brain barrier through inhibiting Aβ(25-35)-induced NLRP3 inflammasome activation via the AMPK/mTOR/ULK1-mediated autophagy in bEnd.3 cells and APP/PS1 mice. Phytother Res. 2021;35(2):954-73 doi:10.1002/ptr.6849
187. Chu D, Dong X, Shi X, Zhang C, Wang Z. Neutrophil-Based Drug Delivery Systems. Adv Mater. 2018;30(22):1706245 doi:10.1002/adma.201706245
188. Liu Y, Xue X, Zhang H, Che X, Luo J, Wang P. et al. Neuronal-targeted TFEB rescues dysfunction of the autophagy-lysosomal pathway and alleviates ischemic injury in permanent cerebral ischemia. Autophagy. 2018Oct18;15(3):493-509 doi:10.1080/15548627.2018.1531196 PubMed PMID: 30304977; PubMed Central PMCID: PMC6351122
189. Cassano T, Magini A, Giovagnoli S, Polchi A, Calcagnini S, Pace L. et al. Early intrathecal infusion of everolimus restores cognitive function and mood in a murine model of Alzheimer's disease. Exp Neurol. 2019Jan1;311:88-105 doi:10.1016/j.expneurol.2018.09.011
190. Els Th, Bruckmann J, Röhn G, Daffertshofer M, Mönting JS, Ernestus RI. et al. Spermidine: A Predictor for Neurological Outcome and Infarct Size in Focal Cerebral Ischemia? Stroke. 2001Jan;32(1):43-6 doi:10.1161/01.STR.32.1.43
191. Satarker S, Wilson J, Kolathur KK, Mudgal J, Lewis SA, Arora D. et al. Spermidine as an epigenetic regulator of autophagy in neurodegenerative disorders. Eur J Pharmacol. 2024Sep15;979:176823 doi:10.1016/j.ejphar.2024.176823
192. Noro T, Namekata K, Kimura A, Guo X, Azuchi Y, Harada C. et al. Spermidine promotes retinal ganglion cell survival and optic nerve regeneration in adult mice following optic nerve injury. Cell Death Dis. 2015Apr;6(4):e1720 doi:10.1038/cddis.2015.93 PubMed PMID: 25880087; PubMed Central PMCID: PMC4650557
193. Liu W, Zhu S ou, Guo Y lin, Tu L fang, Zhen Y qi, Zhao R yan. et al. BL-918, a small-molecule activator of ULK1, induces cytoprotective autophagy for amyotrophic lateral sclerosis therapy. Acta Pharmacol Sin. 2023Mar;44(3):524-37 doi:10.1038/s41401-022-00972-w PubMed PMID: 36042292; PubMed Central PMCID: PMC9958028
194. Egan DF, Chun MGH, Vamos M, Zou H, Rong J, Miller CJ. et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell. 2015Jul16;59(2):285-97 doi:10.1016/j.molcel.2015.05.031
195. Muraleedharan R, Dasgupta B. AMPK in the brain: its roles in glucose and neural metabolism. FEBS J. 2022;289(8):2247-62 doi:10.1111/febs.16151
196. Zhao Y, Zhao W, Han Y. Inhibition of mTORC2 improves brain injury in epileptic rats by promoting chaperone-mediated autophagy. Epilepsy Res. 2023Jul;193:107161 doi:10.1016/j.eplepsyres.2023.107161
197. da Costa A, Metais T, Mouthon F, Kerkovich D, Charvériat M. Evaluating and modulating TFEB in the control of autophagy: toward new treatments in CNS disorders. Fundam Clin Pharmacol. 2021;35(3):539-51 doi:10.1111/fcp.12634
198. Daci R, Flotte TR. Delivery of Adeno-Associated Virus Vectors to the Central Nervous System for Correction of Single Gene Disorders. Int J Mol Sci. 2024Jan15;25(2):1050 doi:10.3390/ijms25021050
199. Esteves M, Cristóvão AC, Vale A, Machado-Pereira M, Ferreira R, Bernardino L. MicroRNA-124-3p Modulates Alpha-Synuclein Expression Levels in a Paraquat-Induced in vivo Model for Parkinson's Disease. Neurochem Res. 2024;49(7):1677-86 doi:10.1007/s11064-024-04130-y PubMed PMID: 38451434; PubMed Central PMCID: PMC11144150
200. Yao L, Ye Y, Mao H, Lu F, He X, Lu G. et al. MicroRNA-124 regulates the expression of MEKK3 in the inflammatory pathogenesis of Parkinson's disease. J Neuroinflammation. 2018Jan12;15:13 doi:10.1186/s12974-018-1053-4 PubMed PMID: 29329581; PubMed Central PMCID: PMC5767033
201. Cignarella F, Filipello F, Bollman B, Cantoni C, Locca A, Mikesell R. et al. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol (Berl). 2020;140(4):513-34 doi:10.1007/s00401-020-02193-z PubMed PMID: 32772264; PubMed Central PMCID: PMC7498497
202. Bayat M, Nahand JS. Let's make it personal: CRISPR tools in manipulating cell death pathways for cancer treatment. Cell Biol Toxicol. 2024Jul29;40(1):1-31 doi:10.1007/s10565-024-09907-z
203. Bhattacharya A, Parillon X, Zeng S, Han S, Eissa NT. Deficiency of Autophagy in Dendritic Cells Protects against Experimental Autoimmune Encephalomyelitis. J Biol Chem. 2014Sep19;289(38):26525-32 doi:10.1074/jbc.M114.575860 PubMed PMID: 25077962; PubMed Central PMCID: PMC4176242
204. Delaney SL, Gendreau JL, D'Souza M, Feng AY, Ho AL. Optogenetic Modulation for the Treatment of Traumatic Brain Injury. Stem Cells Dev. 2020Feb15;29(4):187-97 doi:10.1089/scd.2019.0187
205. Regner A, Meirelles L da S, Simon D, Regner A, Meirelles L da S, Simon D. Traumatic Penumbra: Opportunities for Neuroprotective and Neurorestorative Processes. In. IntechOpen. 2017
206. Loureiro JA, Gomes B, Fricker G, Coelho MAN, Rocha S, Pereira MC. Cellular uptake of PLGA nanoparticles targeted with anti-amyloid and anti-transferrin receptor antibodies for Alzheimer's disease treatment. Colloids Surf B Biointerfaces. 2016Sep1;145:8-13 doi:10.1016/j.colsurfb.2016.04.041
207. Singh D, Kurmi BD, Singh A. Graphene Oxide, a Prominent Nanocarrier to Reduce the Toxicity of Alzheimer's Proteins: A Revolution in Treatment. Recent Pat Nanotechnol. 2025Dec;19(4):572-80 doi:10.2174/0118722105292940240502114430
208. Liu J, Wang T, Dong J, Lu Y. The blood-brain barriers: novel nanocarriers for central nervous system diseases. J Nanobiotechnology. 2025Feb26;23(1):146 doi:10.1186/s12951-025-03247-8
209. Varma VR, Desai RJ, Navakkode S, Wong LW, Anerillas C, Loeffler T. et al. Hydroxychloroquine lowers Alzheimer's disease and related dementias risk and rescues molecular phenotypes related to Alzheimer's disease. Mol Psychiatry. 2023;28(3):1312-26 doi:10.1038/s41380-022-01912-0 PubMed PMID: 36577843; PubMed Central PMCID: PMC10005941
210. Abo-El-Matty D, Rizk M, Aly H, Abd-Alla H, Saleh SM, Younis EA. Role of Fruit Waste and Flavanones-Loaded Silica Nanoparticles in Ameliorating Oxidative Stress and Histological Changes in Rat Brain Induced by Acrylamide. Vol. 9. 2018;9(6):1817-28
211. Murugadoss S, Lison D, Godderis L, Van Den Brule S, Mast J, Brassinne F. et al. Toxicology of silica nanoparticles: an update. Arch Toxicol. 2017Jun1;91(9):2967-3010 doi:10.1007/s00204-017-1993-y
212. Boland B, Yu WH, Corti O, Mollereau B, Henriques A, Bezard E. et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2018Sep;17(9):660-88 doi:10.1038/nrd.2018.109
Author contact
![]() Corresponding authors: Jun Yu; Department of Neurosurgery, Second Affiliated Hospital, School of Medicine, Zhejiang University, 88 Jiefang Rd, Hangzhou, Zhejiang 310009, China. E-mail: 2505020edu.cn. Weilin Xu, Department of Neurosurgery, Second Affiliated Hospital, School of Medicine, Zhejiang University, 88 Jiefang Rd, Hangzhou, Zhejiang 310009, China. E-mail: 11618330edu.cn.
Corresponding authors: Jun Yu; Department of Neurosurgery, Second Affiliated Hospital, School of Medicine, Zhejiang University, 88 Jiefang Rd, Hangzhou, Zhejiang 310009, China. E-mail: 2505020edu.cn. Weilin Xu, Department of Neurosurgery, Second Affiliated Hospital, School of Medicine, Zhejiang University, 88 Jiefang Rd, Hangzhou, Zhejiang 310009, China. E-mail: 11618330edu.cn.