Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusion
Abbreviation
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(12):1461-1471. doi:10.7150/ijbs.16864 This issue Cite
Research Paper
Autophagy activation protects shock wave induced renal tubular epithelial cell apoptosis may through modulation of Akt/ GSK-3β pathway
Qingzhi Long, Xiang Li ![]() , Hui He, Dalin He
, Hui He, Dalin He
Department of Urology, First affiliated hospital, Medical School of Xi'an Jiaotong University, Xi'an, Shaanxi Province, China.
Received 2016-7-15; Accepted 2016-9-17; Published 2016-11-24
Abstract

Purpose: Extracorporeal shock wave lithotripsy (ESWL) is well documented to exert destructive effect to renal cells and its mechanism is not clear. Autophagy is one of cell basic response for stressful conditions and it is important to determine cell's fate. The aim of this study is to elucidate the role of autophagy in the process of shock wave-induced renal cells injury.
Methods: NRK-52E cell, a rat renal tubular epithelial cell, was exposed to shock wave at the voltage of 14KV. GFP-LC3 puncta was used to monitor Autophagy flux in the process of shock wave injury. Autophagic relative proteins, such as light chain 3 (LC3), beclin-1 and p62, were also examined. Cell variability and apoptosis were detected when inhibition autophagy with 3-methyladenine (3MA) or stimulating its activity with rapamycin during the process of shock wave injury. The role of Akt/ GSK-3β and its connection with autophagy in the process of shock wave injury were also investigated.
Results: Shock wave was confirmed to activate autophagy in renal cells, which was manifested in LC3-II turnover, beclin-1 induction and degradation of p62. Inhibition autophagy enhanced cell damage or apoptosis, whereas its stimulating was able to exert protection from shock wave injury. Akt/ GSK-3β, a cell-survival signaling pathway, can also be activated during the process. And its activation could be suppressed by blockade autophagy.
Conclusion: Autophagy is a self-protective response for renal cells from shock wave injury. The cyto-protection of autophagy may be connected with modulation Akt/ GSK-3β pathway.
Keywords: shock wave, autophagy, renal tubular epithelial cell, apoptosis, Akt/ GSK-3β.
Introduction
Extracorporeal shock wave lithotripsy (ESWL) has been extensively used in clinical treatment of upper urinary tract stones. It is well documented that shock wave elicits renal damage and causes several short-term or long-term complications [1-3]. Many clinical trials and animal models have been used to investigate the potential mechanism of shock wave injury to renal cells [4, 5]. Currently the molecular mechanism of shock wave injury remains not clear.
Autophagy is a cellular process that is mainly responsible to digest and recycle long lived proteins or cellular organelles to maintain cell homeostasis. It can be activated under variety of conditions such as hypoxia, oxidative stress, starvation, and toxic reactions. Furthermore, autophagy is proved to play important role in many pathological processes such as tumor [6], neurodegeneration [7], inflammation [8] and ischemia-reperfusion injury [9]. It is controversial whether autophagy exerts survival or destructive effect to cells. Some studies indicated cytoprotective role of autophagy [10, 11] and others drew the opposite conclusion [12]. Thus, the common view is that the role of autophagy to cells fate is diverse and depends on different cell types and conditions.
Previous studies indicated that shock wave-inflicted renal damage is not only the simple mechanical trauma. Many other biological factors such as oxygen free radicals, inflammation and calcium overload are also involved in this process [13-15]. And those factors theoretically can stimulate autophagy process. However, so far there is no report to study the role of autophagy in the process of shock wave injury. We addressed this issue by examining what's the role of autophagy in shock wave-induced renal cell apoptosis, and investigating the possible mechanism that how autophagy determines cell's fate in the process of shock wave induced cell apoptosis.
Materials and methods
1. Cell culture and reagents
NRK52e cell (ATCC, USA), rat renal tubular epithelial cell, was cultured in DMEM with 5% FBS at 37°C in 5% CO2. The chemicals required in this experiment include 3-methyladenine (3MA) and Rapamycin those were purchased from Sigma Aldrich Co (Germany). All of the primary antibodies were purchased from Cell signaling technology (CST, USA). Those antibodies include rabbit anti LC3-I/II, rabbit anti-p62, rabbit anti beclin-1, rabbit anti-cleaved Caspase-3, rabbit anti-Akt, rabbit anti-P-Ser-473-Akt, rabbit anti-GSK-3β, rabbit anti-P-Ser-9-GSK-3β and rabbit anti Atg 5.
2. Experimental model
This study employed the model of renal tubular epithelial cell exposure to shock wave as previously reported [13]. Cultured NRK-52E cells were collected following trypsinization by Trypsin/EDTA digestion and centrifugation for 5 minutes. Cells were suspended in culture medium and prepared for shock wave exposure according to the methods as we have described previously [13]. The shock waves were generated by the HK-MZ-V Lithotripter (Huikang, China), with impulse rates of 50, 100, 150,200,250 and 300 shock waves per minute at a voltage of 14 kV.
3. Cell variability and apoptosis assays
Cell viability was determined by the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay (sigma, Germany). Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) method (Invitrogen, USA) was used for detection of apoptosis at single cell level, based on labeling of free 3'-OH terminal in DNA strand breaks. The cells were washed with PBS and then fixed with 4% paraformaldehyde and stained with Hoechst 33258 (5μg/ml). The stained cells on slides were examined with a fluorescent microscope and photographed with a CCD camera (Olympus, Japan). Cells are determined as apoptotic based on their nuclei present chromatin condensation or marginalization and nuclear beading.
4. Plasmid and small interference RNA transfection
NRK-52E cells were transfected with adenovirus harboring tandem fluorescent GFP-LC3 plasmid (Addgene, USA) with Lipofectamine 3000 according to the manufacturer's (Invitrogen, USA) instructions. After transfection for 24 hours, cells were treated with shock wave 200 impulses at voltage of 14kV. After incubation for another 24 hours in chambers, the sample was fixed and photographed with a fluorescent microscope. Number of autophagosomes (green dots) in the sample was calculated with the Image J software (1.50b NIH).
Small interfering RNA (siRNA) for beclin-1 and Atg 5(CST, USA) was transfected to cells with the lipoinfectamine RNAiMAX (Invitrogen, USA) according to the manufacturer's instruction. After transfection for 24 hours, cells were exposured to shock wave 200 impulses at voltage of 14kV for western blot and apoptosis analysis.
5. Western blot analysis
The protocol of Western blotting used in this study has been previously reported. Briefly, proteins of whole cell lysates (30 µg) were separated by SDS-PAGE and transferred to PVDF membrane. After an overnight blocking with 5% milk in TBST at 4°C, the membrane was incubated with primary antibodies overnight 4°C and then incubated with appropriate horseradish peroxidase-conjugated secondary antibody for 1 hour. Bound antibodies were visualized by chemiluminescence detection.
6. Statistical analysis
Data were expressed as mean ± standard error of the mean and were subjected to ONE WAY ANOVA. Western blotting results were scanned and analyzed for signal intensity with the Image J 1.50b software. Statistical analysis of data was performed using the SPSS 20.0 software. P value was considered as statistically significant if it is less than 0.05.
Results
1. Shock wave decreased viability and caused apoptosis in NRK-52E cells
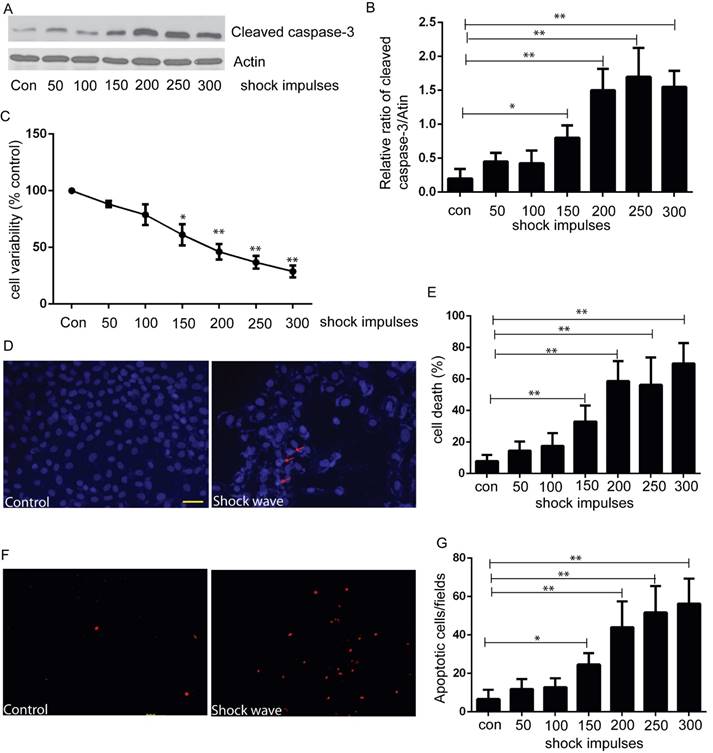
We used MTT assay to estimate cell viability under the condition of shock wave treatment. After exposure to shock wave, cell viability was declined significantly and more serious cell damage was followed with the increase of shock wave impulses (Fig 1C). We then employed western blot to determine the level of cleaved caspas-3, a sensitive apoptosis marker [16]. Cleaved form of caspase-3 was increased significantly due to shock wave treatment (Fig 1A). Furthermore, densitometry quantification of the western blots revealed a significant elevation of cleaved caspas-3 followed with the increase of shock wave impulses (Fig 1B). We also used TUNEL assay combined with nucli staining to estimate the degree of shock wave induced cell apoptosis. More serious apoptotic cell death was observed when it exerted more shock wave impulses for renal cells (Fig 1E to G). These results suggested that shock wave injury could affect viability by activating apoptotic cell death signaling pathway in the subject NRK-52E cells.
shock wave caused NRK52e cell damage and apoptosis. (A): Western blot image of cleaved caspase-3 for NRK52e cell after exposure to shock wave from 50 impulses to 300 impulses. (B): Densitometry was performed for quantification and the ratio of cleaved caspase-3 to actin was expressed as fold of control. Western blots were performed for at least 3 times independently. The relative abundance of cleavage Caspase-3 was elevated significantly followed with increase of shock wave impulses. (C): MTT assay indicated significant decrease of cell variability followed with increase of shock wave impulses. (D): representative images of cell death by using nuclear staining with Hochest 33258. Ten random fields of each picture at 100 × magnifications were analysized (E): Nuclear staining quantitative analysis showed that the ratio of death cell was increased followed with increase of shock wave impulses. (F): representative images of apoptosis positive signals by TUNEL assay which indicated significant increase of apoptotic cell death followed with increase of shock wave impulses. Ten random fields of each picture at 100 × magnifications were analysized. Scale bar is 50um. * p<0.05, ** p<0.01.
2. Shock wave elicited autophagy response in NRK-52E cells
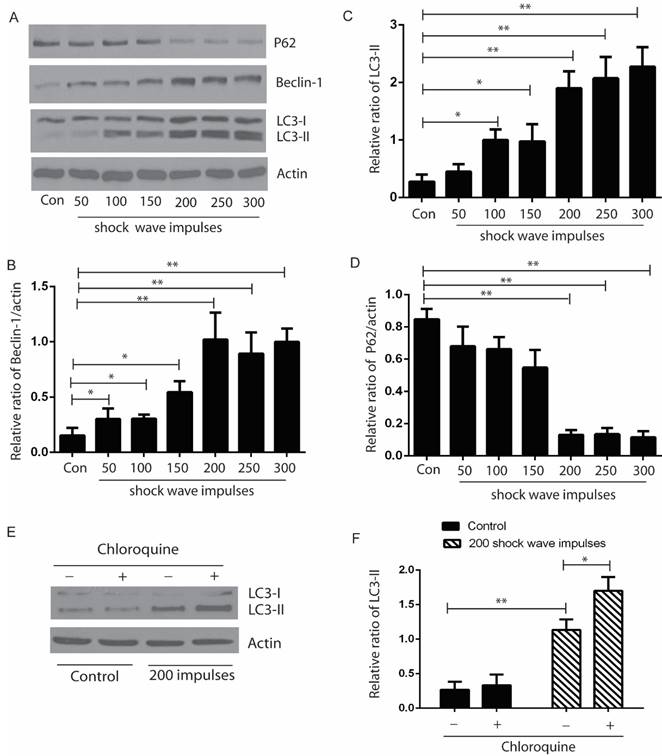
After exposure to shock wave 50 to 300 impulses at the voltage of 14 kV, cells were harvested to examine the autophagic proteins expression by western blots. Our data indicated that the ratio of LC3-II/I and expression of beclin-1 were elevated significantly, and appeared correlating to the increase of shock wave impulses, except that when shock wave treatment was beyond 200 impulses, the ratio of LC-3II/I and beclin-1 expression plateaued. We also found significant degradation of p62 when shock wave treatment beyond 200 impulses. LC3-II turnover was considered to be a principle method to monitor autophagy flux. We thus examined the elevation of LC3-II induced by shockwave can be enhanced by chloroquine (CQ), one of lysosomes inhibitor (Fig 2).
Shock wave triggered autophagy response by examine autophagic proteins expression. The cells were undergoing 200 shock wave impulses with the voltage of 14kv. After treated with shock wave 24 hours' cells were harvested for western blot analysis. A: Representative western blot images of LC3-I/II, p62 and beclin-1. (B-D): Densitometry indicated that there was significant increase of the conversion of LC3-I to LC3-II and beclin-1 and marked decline of P62 expression followed with shock wave impulses increase. E: LC3-II turnover was examined by adding lysosomes inhibitor CQ. F: Densitometry indicated that the increase of LC3-II induced by shockwave was further enhanced by adding CQ. All the experiments were performed for at least 3 times independently. * p<0.05, ** p<0.01.
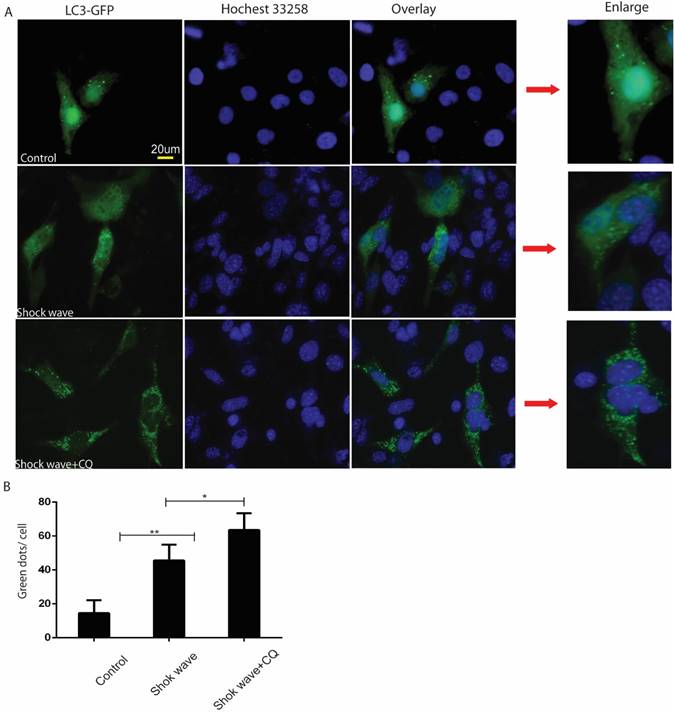
GFP-LC3 puncta analysis was also used to monitor autophagy flux in the process of shock wave injury. In the current study shock wave triggered a significant increase of green GFP-LC3 dots, which represent increase of autophagosomes formation. Those green dots were further elevated by using lysosomes inhibitor CQ, which represents increase of autophagy flux during this process (Fig 3).
Shock wave increased autophagy flux in renal cells. Before exposure to shock wave treatment 24 hours, GFP-LC3 construct was transfused to NRK52e cells by using Lipo3000 system according to the manufactures instruction. The cells were undergoing 200 shock wave impulses with the voltage of 14kv. After treated with shock wave 24 hours' cells were harvested for puncta analysis. LC3-II turnover was examined by using lysosomes inhibitor chloroquine (CQ). A: Representative images of NRK52e cells were transfused GFP-LC3 and exposured to shock wave (200 impulses at the voltage of 14 Kv). Hochest 33258 was used to stain nuclear. Green dots indicate autophagosomes. Random twenty cells of each group at 400×magnification were analysized to examine autophagy flux. B: Quantitative analysis showed that green dots were increased significantly after exposure to shock wave and this increase was enhanced markedly by using lysosomes inhibitor CQ. * p<0.05, ** p<0.01, Scale bar is 20um.
3. Autophagy activation protected shock wave caused renal cell apoptosis
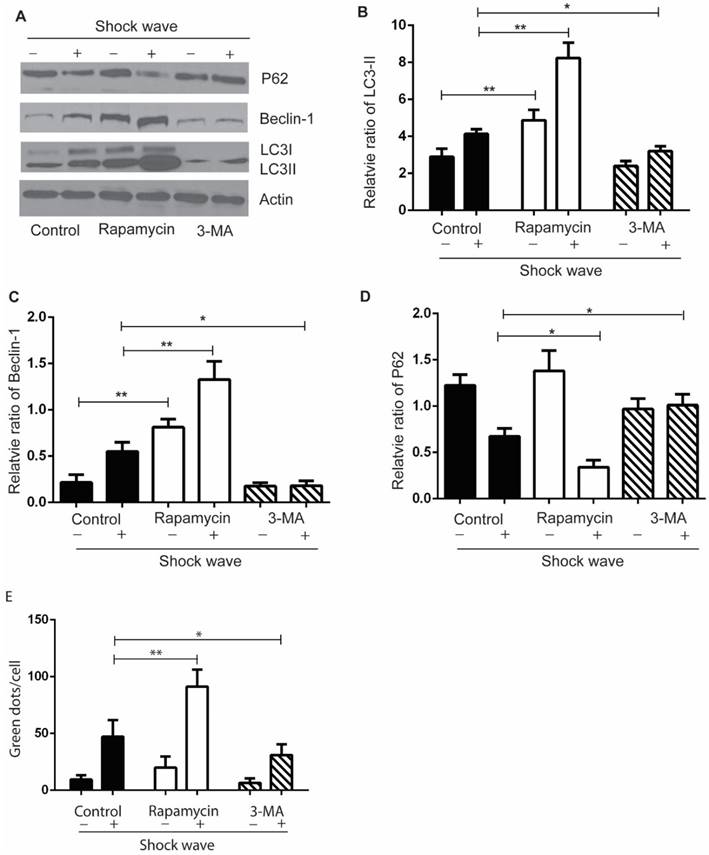
We used autophagy inhibitor, 3-methyladenine (3-MA) [17], to observe the effect of autophagy on cell variability and apoptosis. Cells were pre-treated with 3-MA (5 mg/ml) for 4 hours before shock wave treatment. After 24 hours' incubation, cells were subjected to MTT, TUNEL and nucli staining assay. And cell lysates were detected for the expression of LC3-I/II, beclin-1 and p62 by western blots. The conversion of LC3-II to LC3-1 and beclin-1 expression were found to be declined by 3-MA which also inhibited the degradation of p62, a substrate of autophagy (Fig 4). We next examined that inhibition autophagy with 3-MA caused obvious elevation of caspas-3 cleavage and serious cell damage or apoptosis in the process of shock wave injury (Fig 5).
The effect of rapamycin and 3-MA on shock wave triggered autophagy activation in renal cells. Before exposure to shock wave, NRK52e cells were added with 3-MA (5mg/ml) or rapamycin (200umom/L) for 4 hours. Those chemicals were dissolved with DMSO at the final concentration of 5mg/ml for 3-MA or 200umol/L for rapamycin and control group was only added DMSO. After shock wave 24 hours, cells were harvested to detect autophagy activation. (A) Representative images of western blot for LC3-I/LC3-II, beclin-1 and p62. (B-D) Densitometry was performed for quantification and the ratio of LC3-II, beclin-1 and P62 to actin was expressed as fold of control. All the experiments were performed for at least 3 times independently. * p<0.05, ** p<0.01. The western blots analysis indicated that rapamycin can significantly enhance autophagy activation during the process of shock wave treating cells. On the other hand, 3-MA was able to inhibit LC3-II conversion and suppress beclin-1 expression. The degradation of p62 was also inhibited by using 3-MA during this process. (E): LC-3 GFP puncta analysis indicated that rapamycin could enhance shock wave increased LC-3 puncta, while 3-MA had opposite effect for puncta formation.
Autophagy activation protects renal cell apoptosis induced by shock wave. Before exposure to shock wave, NRK52e cells were added with 3-MA (5mg/ml) or rapamycin (200umom/L) for 4 hours. Those chemicals were dissolved with DMSO at the final concentration of 5mg/ml for 3-MA or 200umol/L for rapamycin and control group was only added DMSO. After shock wave 24 hours' cells were harvested to examine variability and apoptosis. A: Representative western blot images of cleaved Caspase-3. B: Densitometry was performed to indicate that the increase of cleaved caspase-3 was enhanced significantly by using 3-MA. And its elevation could be blocked by using rapamycin. C: Representative images of TUNEL and nucli staining during the process of shock wave treating cells. Red arrow indicates apoptotic cell death. (D and E): TUNEL assay combined with nucli staining by Hochest 33258 indicated that 3-MA enhanced apoptotic cell death while rapamycin was able to protect cell apoptosis. Ten random fields of each group were quantified and analysized at 100×magnification. F: Rapamycin was able to improve cell variability which damaged by shock wave and 3-MA had opposite effect during this process. *P < 0.05, ** P < 0.01.
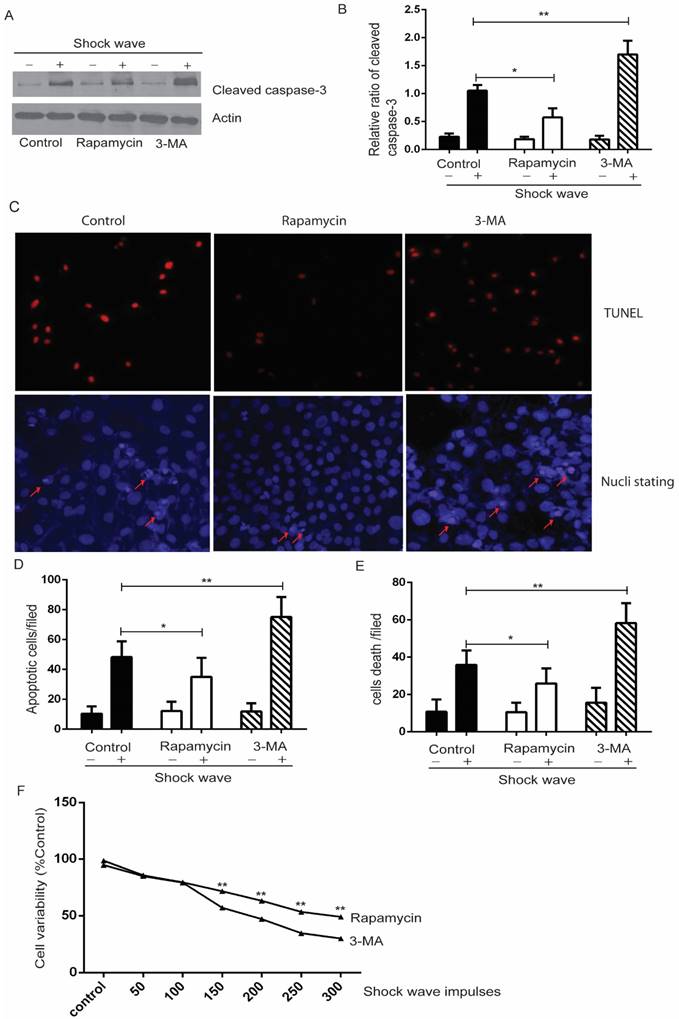
In a complementary experiment, rapamycin, an mTOR inhibitor with the potential of activating autophagy activity [18], was used to confirm the protective effect of autophagy with a similar experimental strategy. Cells were pre-treated with rapamycin (200 µM) for 4 hours before shock wave treatment. The results indicated that rapamycin-induced autophagy activation (Fig 4) significantly decreased the level of cleaved Caspase-3 and numbers of apoptotic cells (Fig 5).
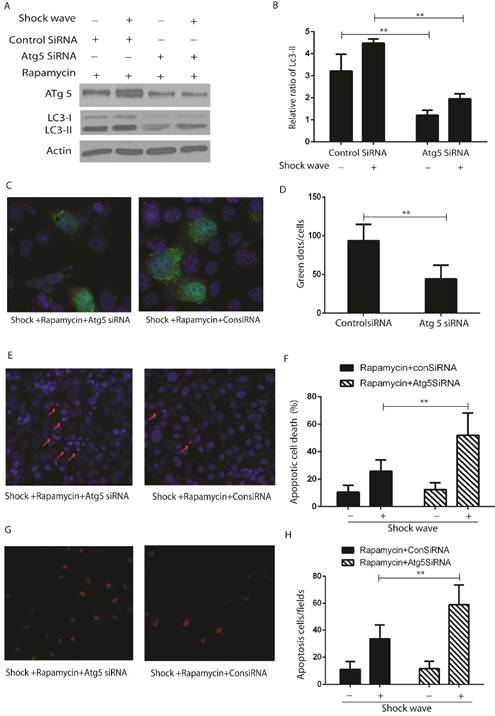
4. The effect of rapamycin to protect shock wave induced cell apoptosis is due to induct autophagy activation
LC3-II western blot (Fig 6 A-B) and GFP-LC3 puncta analysis (Fig 6 C-D) indicated that activation of autophagy with rapamycin can be suppressed by knockdown Atg 5, which is a key gene to induct and keep autophagy flux, in the process of shock wave treating cells. Nucli staining (Fig 6 E-F) and TUENL assay (Fig G-H) confirmed that inhibition autophagy by knockdown Atg 5 could block the protection of rapamycin in the process of shock wave induced cell apoptosis.
The protective effect of autophagy was confirmed by knockdown Atg5 in the process of shockwave injury renal cells. (A-B): Western blot indicated that knockdown Atg5 can significantly block LC3-II conversion. (C-D): Knockdown Atg5 made a significant decline of LC3-GFP green dots in the process of shock wave and rapamycin treating NRK52e cells. Twenty random cells of each picture at 400 × magnifications were analysized. (E-F): Nucli staining indicated that there was more serious apoptotic death in NRK52e cells when knockdown Atg5. (G-H): More apoptosis was found in Atg5 knockdown cells by TUNEL assay. Ten random fields of each picture at 100 × magnifications were analysized.
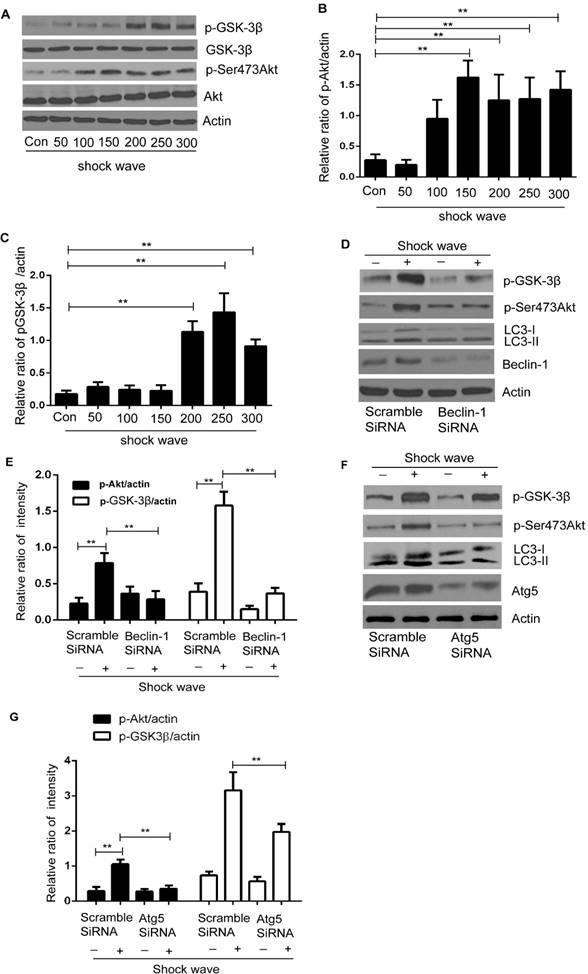
5. Autophagy is able to modulate Akt/GSK-3β, which is cell survival signaling pathway, in the process of shock wave induced cell apoptosis
We examined Akt/GSK pathway, one of important cell-survival signaling pathway, to investigate how autophagy protect shock wave induced renal cell apoptosis. A significant increase of phosphorylated Ser473 Akt was detected when shock wave treating beyond 100 impulses. And if it keeps elevating to 200 impulses, we can observe marked increase of phosphorylated GSK-3β, a downstream gene of Akt (Fig 7A-C). We further investigated the possible connection between autophagy and Akt/GSK-3β pathway through knockdown autophagic gene beclin-1 and Atg5. The Akt and GSK-3β phosphorylation can be suppressed by inhibition autophagy with knockdown beclin-1 (Fig 7 D-E) or Atg5 (Fig 7 F-G). We thus concluded that Akt/ GSK-3β, the cell survival signaling pathway, can be activated during the process of shock wave injury. And in this process, autophagy is necessary to maintain and regulate the phosphorylation of Akt/ GSK-3β.
Akt/GSK-3β pathway was activated during the process of shock wave injury renal cells. And the activation of this pathway can be maintained and regulated by autophagy. (A): Representative western blot images of Akt/p-Ser473Akt and GSK-3β/p-GSK-3β (B and C): Densitometry was performed for quantification and the ratio of p-Ser473Akt or p-GSK-3β to actin was expressed as fold of control. The analysis showed that shock wave significantly elevated the expression of p-Akt and p-GSK-3β. (D-E): Knockdown beclin-1 by siRNA both blocked autophagy activation and phosphorylation of Akt/GSK pathway during the process of shock wave treating cells. (F-H): Knockdown Atg5 by siRNA both suppressed autophagy activation and phosphorylated Akt and its downstream gene GSK-3β. All the experiments were performed for at least 3 times independently. *P < 0.05, ** P < 0.01.
Discussion
ESWL is widely used to treat urinary tract stones. This physical treatment has side effect to injury renal tissue. And its mechanism is not clear. Major optioning so far considered that shock wave can exert shear stress, oxygen free radicals, inflammations and other signals to damage renal cells [13-15]. Theatrically those factors are able to activate autophagy activation. The purpose of this study is to examine the role of autophagy in shock wave injury renal cells and its mechanism.
Our experiment detected significant increase of the conversion of LC-3I to LC3-II and beclin-1 expression in renal cells followed with increase of shock wave impulses. The cytosolic LC3-I converts to phospholipid conjugate LC3-II is considered to represent activation of autophagy [19]. Becinl-1 is an import gene to regulate and activate autophagy process. It's up regulation is a sign of autophagy induction in the upstream of autophagic pathway [20]. The degradation of autophagic substrate can also be detectable when autophagy is activated. Our experiment examined obvious degradation of p62, a substrate of autophagy [21], during the process of shock wave treating cells (Fig 2).
We also used GFP-LC3 puncta to monitor autophagy flux in the process. As shown in figure 2, only a few green dots appeared in control cells. However, great deal of green dots was detectable in shock wave treated cells, which indicated significant increase of autophagosomes in those cells [22]. LC3-II turnover is believed to estimate activation of autophagy flux by using lysosomes inhibitors [23]. We next examined that the increase of LC3-II expression (Fig 3 E-F) and puncta (Fig 2), which induced by shock wave, can be further enhanced by chloroquine, a lysosomes inhibitor. These data indicated that more amounts of autophagosomes were produced and delivered to lysosome for degradation during the process of shock wave injury cells. The result of LC3-II turnover, together with autophagic proteins expression and puncta data provide stronger evidence that shock wave is able to activate autophagy flux in renal cells.
It is highly controversial of the role of autophagy in kidney diseases. It may exert cytoprotective or destructive effect depending on cells and stressful conditions. Some study indicated that autophagy exerts cell survival from kidney injury [24]. However, others come out the opposite conclusion that autophagy may trigger more serious cell damage or death [25]. Our experiment was firstly addressed to investigate the role of autophagy in shock wave injury renal cell by suppression or induction autophagy activation. We observed more serious cell damage and apoptosis due to shock wave when autophagy was suppressed by 3-MA, a specific autophagy inhibitor (Fig 5). On the other hand, rapamycin was proved to improve cell variability and alleviate apoptosis during the process of shock wave injury (Fig 5). It is not clear whether rapamycin exerts cell survival is due to induction autophagy because of its multiple function. Thus, we addressed next experiment to focus on this issue by knockdown Atg 5, which is an E3 ubiquitin ligase and necessary for autophagy. Atg 5 is considered to form a complex with Atg 12 and that complex is necessary for LC3-1 conjugation to PE to form LC3-II [26]. Our result indicated that the protection of rapamycin to alleviate shock wave induced renal cell apoptosis can be blocked when autophagy was inactivated by knockdown Atg5 [Fig 6]. This data confirmed the positive effect of autophagy to prevent shock wave induced renal cell apoptosis.
How autophagy works to play cytoprotective effect in the process of shock wave injury? It is well known that several signal transduction pathways can determine cell's fate under variety of stressful conditions [27]. Among those signaling pathways, phosphoinositide 3-kinase (PI3K)/Akt is mainly involved in oxidative stress triggered cell death and is considered to deliver survival signals to protect cells from apoptosis [28]. In the current study, we examined the marked increase of Akt phosphorylation when shock wave treatment beyond 100 impulses and if this treatment beyond 200 impulses the significant increase of phosphorylation of glycogen synthase kinase-3 (GSK-3β) can also be observed (Fig A-C). GSK-3β is one of the apoptosis regulatory molecular and can be phosphorylated by Akt at serine 9. The phosphorylation of GSK-3β is able to reduce its kinase activity and offers cell survival [29]. Thus, activation of Akt/ GSK-3β pathway may be a self-protective response for cells to against shock wave injury.
Is there connection between autophagy and Akt/ GSK-3β pathway during the process of shock wave injury renal cells? We addressed further experiment to examine whether autophagy is able to regulate Akt/ GSK-3β pathway by knockdown autophagy regulatory gene becin-1 and Atg5. Our data confirmed that autophagy is necessary to keep activation of Akt/ GSK-3β in the process of shock wave injury. We observed marked decline of phosphorylation of Akt and GSK-3β when inhibition autophagy by knockdown beclin-1 (Fig 7 D-E) or Atg5 (Fig 7 F-H). Based on this very intriguing result we may conclude that the protection of autophagy for renal cells from shock wave injury is due to regulate Akt/ GSK-3β pathway.
Conclusion
Autophagy is a self-protective cellular response for renal tubular epithelial cells from shock wave injury. The cyto-protection of autophagy may attribute to its ability to regulate Akt/ GSK-3β pathway, which is an important signaling pathway to exert cell survival under oxidative stressful condition. Our experiment is very new and provides a novel research direction to study the molecular mechanism of shock wave lithotripsy caused renal damage. Further study is required to investigate the molecular event of how autophagy regulates Akt/ GSK-3β and other signaling pathways, which can determine cells fate in shock wave injury renal cells.
Abbreviation
Atg5: Autophagy protein 5
Atg 12: Autophagy protein 12
ESWL: Extracorporeal shock wave lithotripsy
LC3: Light chain 3
RTPC: Renal tubular epithelial cells
3MA: 3-methyladenine
MTT assay: 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide assay
TUNEL: Terminal deoxynucleotidyl transferase dUTP nick-end labeling
siRNA: Small interfering RNA
ANOVA: Analysis of Variance
Acknowledgements
This research work was funded by the National Natural Science Foundation of China under Grant No. 30700833.
Competing Interests
The authors declare that they have no conflict of interest.
References
1. Aydin HR, Irkilata L, Aydin M. et al. The Application of kidney injury molecule-1 to determine the duration between shockwave lithotripsy sessions. J Endourol. 2016;30:109-113
2. Lee HY, Yang YH, Shen JT. et al. Risk factors survey for extracorporeal shockwave lithotripsy-induced renal hematoma. J Endourol. 2013;27:763-767
3. Hadj Moussa M, Brown JA. Effect of high shock number on acute complication development after extracorporeal shockwave lithotripsy. J Endourol. 2013;27:1015-1019
4. Jaeger P, Redha F, Marquardt K. et al. Morphological and functional changes in canine kidneys following extracorporeal shock-wave treatment. Urol Int. 1995;54:48-58
5. Skuginna V, Nguyen DP, Seiler R. et al. Does Stepwise Voltage Ramping Protect the Kidney from injury during extracorporeal shockwave lithotripsy? Results of a prospective randomized trial. Eur Urol. 2006;69:267-273
6. Nishikawa M, Miyake H, Liu B. et al. Expression pattern of autophagy-related markers in non-metastatic clear cell renal cell carcinoma: association with disease recurrence following radical nephrectomy. J Cancer Res Clin Oncol. 2015;141:1585-1591
7. Kiriyama Y, Nochi H. The Function of Autophagy in Neurodegenerative Diseases. Int J Mol Sci. 2015;16:26797-26812
8. Chen ZH, Wu YF, Wang PL. et al. Autophagy Is Essential for Ultrafine Particle-Induced Inflammation and Mucus Hyperproduction in Airway Epithelium. Autophagy. 2016;12:297-311
9. Yang J, Wang Y, Sui M. et al. Tri-iodothyronine preconditioning protects against liver ischemia reperfusion injury through the regulation of autophagy by the MEK/ERK/mTORC1 axis. Biochem Biophys Res Commun. 2015;467:704-710
10. Reumann S, Shogren KL, Yaszemski MJ. et al. Maran A. Inhibition of Autophagy Increases 2-Methoxyestradiol-Induced Cytotoxicity in SW1353 Chondrosarcoma Cells. J Cell Biochem. 2016;117:751-759
11. Mei S, Livingston M, Hao J. et al. Autophagy is activated to protect against endotoxic acute kidney injury. Sci Rep. in press. doi: 10.1038/srep22171
12. Horie R, Nakamura O, Yamagami Y. et al. Apoptosis and antitumor effects induced by the combination of an mTOR inhibitor and an autophagy inhibitor in human osteosarcoma MG63 cells. Int J Oncol. 2016;48:37-44
13. Li X, He D, Zhang L. et al. Pyrrolidine dithiocarbamate attenuate shock wave induced MDCK cells injury via inhibiting nuclear factor-kappa B activation. Urol Res. 2007;35:193-199
14. Li X, Xue Y, He D. et al. Shock wave induces chronic renal lesion through activation of the nuclear factor kappa B signaling pathway. World J Urol. 2010;28:657-662
15. Li X, Long Q, Cheng X. et al. Shock wave induces biological renal damage by activating excessive inflammatory responses in rat model. Inflammation. 2014;37:1317-1325
16. Kuboki M, Ito A, Simizu S. et al. Activation of apoptosis by caspase-3-dependent specific RelB cleavage in anticancer agent-treated cancer cells: involvement of positive feedback mechanism. Biochem Biophys Res Commun. 2015;456:810-814
17. Wu Y, Wang X, Guo H. et al. Synthesis and screening of 3-MA derivatives for autophagy inhibitors. Autophagy. 2013;9:595-603
18. Graziotto JJ, Cao K, Collins FS. et al. Rapamycin activates autophagy in Hutchinson-Gilford progeria syndrome: implications for normal aging and age-dependent neurodegenerative disorders. Autophagy. 2012;8:147-151
19. Tanida I, Minematsu-Ikeguchi N, Ueno T. et al. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84-91
20. Maejima Y, Isobe M, Sadoshima J. Regulation of autophagy by Beclin 1 in the heart. J Mol Cell Cardiol. 2016;95:19-25
21. Komatsu M, Ichimura Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 2010;584:1374-1378
22. Kaminskyy V, Abdi A, Zhivotovsky B. A quantitative assay for the monitoring of autophagosome accumulation in different phases of the cell cycle. Autophagy. 2011;7:83-90
23. Loos B, du Toit A, Hofmeyr JH. Defining and measuring autophagosome flux—concept and reality. Autophagy. 2014;10:2087-2096
24. Buchser WJ, Laskow TC, Pavlik PJ. et al. Cell-mediated autophagy promotes cancer cell survival. Cancer Res. 2012;72:2970-2979
25. Armstrong JL, Hill DS, McKee CS. et al. Exploiting cannabinoid-induced cytotoxic autophagy to drive melanoma cell death. J Invest Dermatol. 2015;135:1629-1637
26. Tanida I, Tanida-Miyake E, Nishitani T. et al. Murine Apg12p has a substrate preference for murine Apg7p over three Apg8p homologs. Biochem Biophys Res Commun. 2002;292:256-262
27. Zhuang S, Yan Y, Daubert RA. et al. ERK promotes hydrogen peroxide-induced apoptosis through caspase-3 activation and inhibition of Akt in renal epithelial cells. Am J Physiol Renal Physiol. 2007;292:F440-447
28. de Araujo WM, Robbs BK, Bastos LG. et al. PTEN Over expression Cooperates With Lithium to Reduce the Malignancy and to Increase Cell Death by Apoptosis via PI3K/Akt Suppression in Colorectal Cancer Cells. J Cell Biochem. 2016;117:458-469
29. De Sarno P, Li X, Jope RS. Regulation of Akt and glycogen synthase kinase-3 beta phosphorylation by sodium valproate and lithium. Neuropharmacology. 2002;43:1158-1164
Author contact
![]() Corresponding author: Xiang Li, No.277 Yanta West Road, Xi'an, Shaanxi Province, China (710061). Email: doctor_seancom; mnwklx85323945edu.cn. Tel number: +86-29-8532-3661 Fax number: +86-29-85324058.
Corresponding author: Xiang Li, No.277 Yanta West Road, Xi'an, Shaanxi Province, China (710061). Email: doctor_seancom; mnwklx85323945edu.cn. Tel number: +86-29-8532-3661 Fax number: +86-29-85324058.