Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(12):1533-1543. doi:10.7150/ijbs.15943 This issue Cite
Research Paper
Cyclooxygenase-2 in tumor-associated macrophages promotes metastatic potential of breast cancer cells through Akt pathway
Lu Gan1,2, Zhu Qiu1, Jing Huang1, Yunhai Li1,3, Hongyan Huang1, Tingxiu Xiang1, Jingyuan Wan4, Tianli Hui1, Yong Lin1, Hongzhong Li1,3 ![]() , Guosheng Ren1,3
, Guosheng Ren1,3 ![]()
1. Chongqing Key Laboratory of Molecular Oncology and Epigenetics, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China;
2. Department of Oncology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China;
3. Department of Endocrine and Breast Surgery, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China;
4. Department of Pharmacology, Chongqing Medical University, Chongqing, China.
Received 2016-4-25; Accepted 2016-10-9; Published 2016-11-26
Abstract

Tumor-associated macrophages (TAMs) promote cancer development and progression by releasing various cytokines and chemokines. Previously, we have found that the number of COX-2+ TAMs was associated with lymph node metastasis in breast cancer. However, the mechanism remains enigmatic. In this study, we show that COX-2 in breast TAMs enhances the metastatic potential of breast cancer cells. COX-2 in TAMs induces MMP-9 expression and promotes epithelial-mesenchymal transition (EMT) in breast cancer cells. In addition, COX-2/PGE2 induces IL-6 release in macrophages. Furthermore, we find that the activation of Akt pathway in cancer cells is crucial for the pro-metastatic effect of COX-2+ TAMs by regulating MMP-9 and EMT. These findings indicate that TAMs facilitate breast cancer cell metastasis through COX-2-mediated intercellular communication.
Keywords: Tumor microenvironment, macrophages, breast cancer, cyclooxygenase-2.
Introduction
Tumor associated macrophages (TAMs) are known to be an important component of tumor microenvironment, which promote tumor growth, angiogenesis, and metastasis through releasing various factors for cancer progression [1,2]. Macrophages have a tremendous plasticity and change their functional profiles repeatedly in response to different environmental stimuli. When exposed to lipopolysaccharides and IFN-γ, macrophages are polarized to M1 macrophages. Conversely, when exposed to Th2 cytokines such as IL-4 and IL-13, they are polarized to M2 macrophages. Compared with M1 macrophages, M2 macrophages do not produce proinflammatory mediators such as TNF-α, IL-1β and IL-12/23, but express high levels of immunosuppressive cytokines such as IL-10 and TGF-β, high arginase-1 activity and specific surface markers such as CD163 and CD206. Generally, macrophages at early stages of tumor initiation show an M1 phenotype which exerts strong tumoricidal activities, while TAMs in established tumors show an M2 phenotype that enhances tumor progression and accelerates tumor aggressiveness [3,4].
Cyclooxygenase-2 (COX-2) is the rate-limiting enzyme in the metabolic conversion of arachidonic acid (AA) into various prostaglandins (PGs) including prostaglandin E2 (PGE2). COX-2 over-expression has been found in many malignancies including breast cancer, which contributes to carcinogenesis by inducing cancer cell proliferation, inhibiting apoptosis, and increasing metastasis [5,6]. Furthermore, COX-2 plays an essential role in linking inflammation and immunity to cancer [7,8]. COX-2 over-expression in tumor microenvironment, particularly in macrophages, enhanced tumor progression in melanoma or prostate cancer [9,10]. In our previous study, we showed that COX-2 in TAMs promoted breast cancer cell survival by triggering a positive-feedback loop between macrophages and cancer cells. COX-2 in TAMs induced the expression of COX-2 in breast cancer cells, which in turn promoted M2 macrophage polarization. Moreover, high COX-2 expression in TAMs correlated with poor prognosis in breast cancer patients. The number of COX-2+ TAMs was associated with lymph node metastasis [11]. These findings suggest that COX-2 in TAMs has an important influence on breast cancer metastasis, which needs further investigation. In this study, we continue to investigate the contribution of COX-2 in TAMs to breast cancer metastasis, and to explore the mechanisms underlying the process. The results suggest that TAMs facilitate breast cancer cell metastasis through COX-2-mediated intercellular communication that involves IL-6 secretion from macrophages and activation of the Akt pathway in cancer cells.
Materials and methods
Generation of macrophages or tumor associated macrophages
Mononuclear cells from the blood of healthy donors were incubated in 6-well plates for 2 hours at 37°C to remove non-adherent cells. The adherent monocytes were incubated for 7 days in medium with M-CSF to become normal macrophages (monocyte-derived macrophages, MDMs). MDMs were co-cultured with malignant MDA-MB-231 breast cancer cells for an additional 7 days to generate tumor associated macrophages (TAMs) [12].
Adenovirus infection
The adenovirus expressing empty Ad-Easy1 vector, COX-2, Akt1, scrambled siRNA or siRNA COX-2 or Akt1 was used following the procedure described previously [11]. In addition to the expression of transgenes, the adenovirus expressing system also expressed RFP as a marker for monitoring transfection efficiency. A series of infections using various dilutions of adenovirus were conducted to determine the optimal multiplicity of infection (MOI) in which expression of target genes occurred with low cytotoxicity.
Wound healing assay
The cancer cells were cultured in 6-well plates in medium containing 10% FBS. When the cells were nearly confluent, the cell monolayer in each well was carefully scratched with a plastic pipette tip to create a linear “wound”. The monolayer was washed twice with PBS to remove debris and detached cells, and the cells were then exposed to serum-free medium for 12 h. The wound closure was monitored and photographed using a microscope fitted with a Leica camera. By comparing the images from when the wound was generated to the last time point (12 h), the degree of wound closure was then quantitatively evaluated using Image-Pro Plus software. Four fields from each well were documented, and each experiment was performed in triplicate.
Cell invasion assay
The cancer cell invasion assay was conducted with transwell membranes (8 μm, pore size, 24-well plate, BD Biosciences, Billerica, MA, USA). The transwell membranes were first coated with 100 μl Matrigel matrix (1 mg/ml, BD Biosciences). Cells suspended in serum-free medium were added to the upper wells (20000 cells/well), while migration-inducing medium (with 10% FBS) was added to the lower wells. After 24 h, the top surface of the chambers was scraped clean with a cotton swab. The cells on the lower surface of the membranes were fixed for 15 min with methanol and then stained with Giemsa solution. Completed transmigration was evaluated by microscopy. Four random fields per filter were scanned for the presence of cells on the lower side of the membrane.
Human MMP antibody array
The expression profile of MMP-related proteins was analyzed using a human MMP array kit (AAH-MMP-1, RayBiotech, Inc., Norcross, GA, USA), which contains duplicate spots of 10 MMP-related proteins (Figure 2A). Briefly, each captured antibody was printed on the membrane, and then treated or untreated cell lysate was added to the antibody array membranes. After extensive washing, the membranes were incubated with a cocktail of biotin-conjugated antiapoptotic protein antibodies. After incubation with HRP-streptavidin, the signals were visualized by chemiluminescence. The relative expression levels of target proteins were determined by comparing the signal intensities quantified by densitometry. A positive control was used to normalize the results from the different membranes.
Western blot
Cell lysate was prepared according to the method described by the protein extract kit (Active Motif Company, Carlsbad, USA). Protein concentrations were determined by BCA protein assay kit (Pierce Biotechnology Inc, Rockford, USA). Cell lysate was analyzed for western blot analysis using COX-2 (Cell Signaling Technology, Inc., Danvers, MA, CST #12282), MMP-9 (CST #13667), E-cadherin (CST #3195), Vimentin (CST #5741), N-cadherin (CST #13116), Snail (CST #3879), phosphor(p)-Akt (CST #4060) plus β-actin (Abcam Inc., Cambridge, MA, ab8226). Antibody binding was visualized with an ECL chemiluminescence system and short exposure of the membrane to X-ray films (Kodak, Japan). Densitometric analysis was done using Image Pro-Plus software and normalized to β-actin.
Active human MMP-9 fluorescence assay
MMP-9 activity was quantified using an ELISA-based activity assay (F9M00, R&D systems, Inc., Minneapolis, MN, USA) according to the manufacturer's instructions. Briefly, the cell culture supernatants and standards were incubated for 2 h at room temperature in 96-well plates coated with monoclonal antibodies for MMP-9, then chemically activated with p-aminophenylmercuric acetate. After incubation for another 2h at 37 ℃, a fluorogenic substrate was added to each well, and the plate was incubated at 37 ℃ for 20 h in dark humidified environment. The plate was then read on a spectrophotometer with excitation and emission wavelengths of 320 and 405, respectively. The data were quantified using standard curves generated from reagents provided with the kit.
Indirect immunofluorescence analysis
For the immunofluorescence experiments in cell lines, the cells were prepared and analyzed under a fluorescence microscope (Leica DM IRB) as described previously [13]. Briefly, the cells were incubated with a primary antibody to E-cadherin or Vimentin (Cell Signaling Technology, Inc.) and then with the DyLight 549 or DyLight 488 antirabbit IgG secondary antibody (CWBiotech, Beijing, China). The cells were then counterstained with DAPI and imaged with a fluorescence microscope (Leica DM IRB). For the immunofluorescence experiments in clinical samples, paraffin embedded tissues were prepared and analyzed under fluorescence microscope following the procedure described previously. Briefly, samples were incubated with primary mouse antibody against CD163 and rabbilt antibody against COX-2, and then incubated with DyLight 488 against mouse IgG or DyLight 549 secondary antibody against rabbit IgG (Cwbiotech, Beijing, China). Cells were then counterstained with DAPI and imaged with the fluorescence microscope.
Clinical data collection
Breast cancer tissue microarrays (HBre-Duc170Sur-01, Outdo Biotech Co., Shanghai, China) include primary breast carcinoma samples from 160 patients with a median age of 53 years and a median follow-up period of 118 months at the hospitals in central China, which are the same with our previous study [11]. Every sample dot with a diameter of 1.5 mm and a thickness of 4 µm was prepared according to a standard method. All samples were collected with informed consent from patients, and all related procedures were performed with the approval of ethics boards of the indicated hospitals.
EIA
The levels of PGE2 in macrophages were determined by EIA using human EIA Kits, according to the manufacturers' instruction (Cayman Chemicals Co., Ann Arbor, MI, USA).
ELISA
The levels of IL-6 in macrophages were determined by ELISA using human ELISA Kits, according to the manufacturers' instruction (R&D Systems, Minneapolis, MN, USA).
Animal experiments
All the animal studies were approved by the Animal Ethics Committee of Chongqing Medical University. 5-Week old severe combined immunodeficiency (SCID) hairless female mice were purchased (Institute of Laboratory Animal Science, Chinese Academy of Medical Science, Beijing, China) and randomly divided into four groups of 10 mice each. All the mice were housed according to the national and institutional guidelines for humane animal care. Macrophages in mice were depleted by injection of freshly prepared clodronate-containing liposomes as described [14]. At 6 weeks of age, the mice were injected subcutaneously on the right rear flanks with 4T1 cells that were prior admixed with COX-2 over-expression, COX-2 knockdown or wild-type RAW264.7-derived TAMs, respectively. Body weights were monitored weekly as an indicator of overall health. After 4 weeks, the mice were euthanized via CO2 asphyxiation. Tumors were then removed, weighed, and sent for immunohistochemistry (IHC) analysis.
IHC
The same tumor tissues from our previous study [11] were fixed in 4% formaldehyde solution (pH 7.0) and subsequently embedded in paraffin. Immunohistochemical studies were performed using the standard streptavidin-peroxidase (SP) method with the UltraSensitive TM SP Kit (Maixin-Bio, Fujian, China) according to the manufacturer's instructions. Tumor specimens were stained using MMP-9, E-cadherin or Vimentin antibodies. Negative control was performed by replacing the primary antibody with PBS. Immunostained slides were blindly evaluated by a trained pathologist under a transmission light microscope.
Statistical analysis
All statistical analysis was done using SPSS 18.0 software. The data in cell experiments were presented as the mean values±standard deviation (SD). All statistical analysis was performed using the Student's t-test or one-way ANOVA followed by Tukey post hoc test. Pearson's correlation and regression analysis was performed to assess the relationship between COX-2+ TAMs and Vimentin in the enrolled samples. Differences were considered significant when the p values were 0.05.
Results
High COX-2 expression in TAMs promotes breast cancer cell migration and invasion
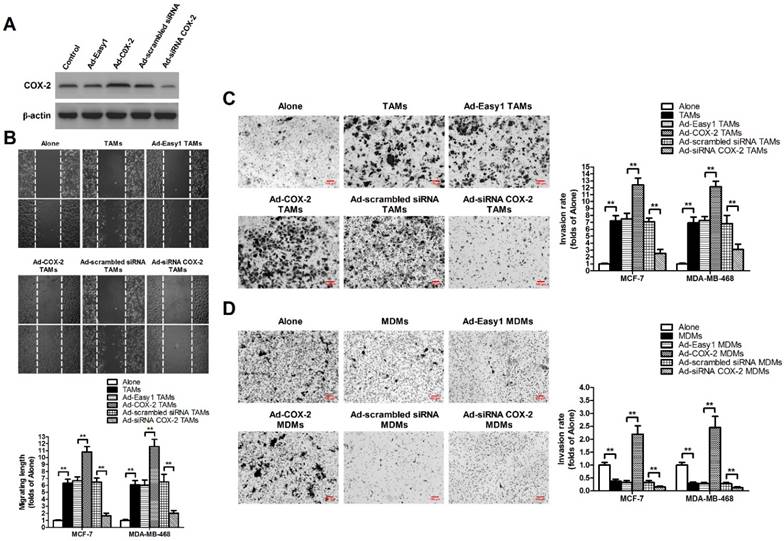
TAMs were established by in vitro co-culture of normal monocyte-derived macrophages (MDMs) with breast cancer cells for 7 days [11]. In order to evaluate the effect of COX-2 in breast TAMs on the metastatic potential of breast cancer cells, TAMs were first transfected with adenoviral COX-2 or siRNA COX-2 (Figure 1A), and then co-cultured with different breast cancer cell lines (MCF-7 and MDA-MB-468) for 7 days. The metastatic potential of breast cancer cells were measured by wound-healing assays and transwell invasion assays. We found that TAMs promoted the metastatic potential of cancer cells, which was enhanced by COX-2 over-expression, but attenuated by COX-2 knockdown in TAMs (Figure 1B-1C). Furthermore, it was also shown that over-expression of COX-2 in MDMs promoted the invasive ability of breast cancer cells while normal MDMs inhibited it, suggesting COX-2 might reverse the antitumor activity of MDMs (Figure 1D).
COX-2 in TAMs promotes migration and invasion of breast cancer cells. (A) The expression of COX-2 in TAMs transfected with adenoviral COX-2 or siRNA COX-2 was checked by western blot. β-actin was used as an internal loading control. The blots shown are representative of six independent experiments. (B) For the wound healing assay, confluent monolayers of MCF-7 and MDA-MB-468 cells co-cultured with or without (Alone) TAMs transfected with adenoviral COX-2 or siRNA COX-2 for 7 days were scarred. The migration of the cells into the wound zone was monitored by microscopy over a 12-h period. The upper panel shows the wound healing assay in MCF-7 cells. White lines indicate the scraped zone. (C) and (D) The transwell invasion assay. After MCF-7 and MDA-MB-468 cells were co-cultured with or without (Alone) TAMs (C) or MDMs (D) transfected with adenoviral COX-2 or siRNA COX-2 for 7 days, the cells that migrated to the lower chamber or invaded through the Matrigel were fixed, stained, and counted using a light microscope. Four random fields per filter were scanned for the presence of cells on the lower side of the membrane. The upper panel shows the cell invasion assay in MCF-7 cells (original magnification, ×100). All experiments were performed thrice in triplicate. The data are presented as the mean ± SD. *p < 0.05 and **p < 0.01 (versus Alone group).
COX-2 in TAMs regulates MMP-9 expression in breast cancer cells
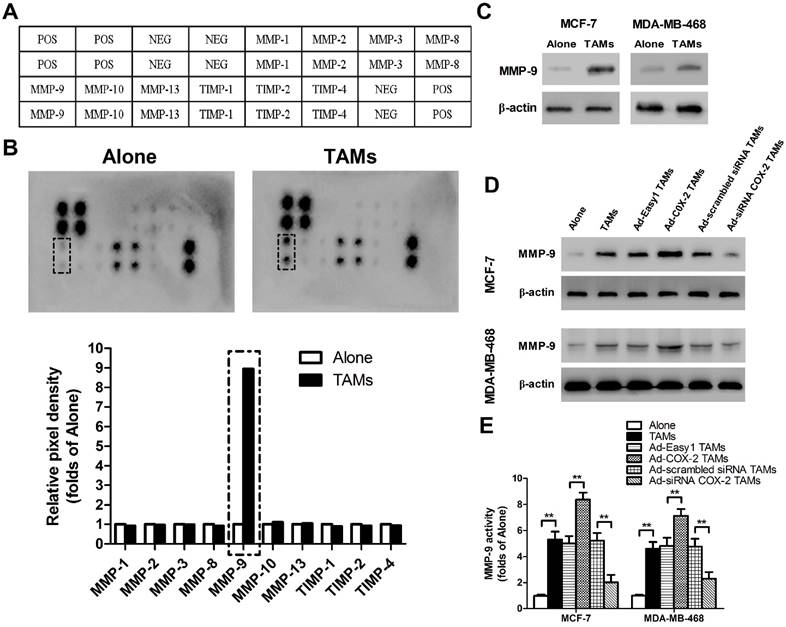
Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs), which function in remodeling the extracellular matrix (ECM), are closely implicated in tumor invasion and metastasis [15]. To elucidate whether or which MMPs/TIMPs were involved in TAMs-mediated cancer cell migration and invasion, human MMPs arrays were performed in MCF-7 breast cancer cells (Figure 2A). As shown in Figure 2B, MMP-9 was significantly up-regulated by TAMs, which was also confirmed by Western blot. Similarly, increased MMP-9 expression and activity were observed in MDA-MB-468 cells co-cultured with TAMs (Figure 2C-2E). Furthermore, COX-2 over-expression in TAMs enhanced MMP-9 expression in breast cancer cells, while inhibiting COX-2 by siRNA suppressed this effect (Figure 2D-2E).
COX-2 in TAMs induces MMP-9 expression in breast cancer cells. (A) Template showing the location of the MMP/TIMP antibodies spotted onto the RayBio Human MMP Array Kit. POS: positive; NEG: negative. (B) TAMs-induced modulation of MMP/TIMP proteins in MCF-7 cells. After MCF-7 cells were co-cultured with or without TAMs for 7 days, the cell lysate was applied to the antibody array. The pixel density was measured, and the data are presented as ratios (compared to the Alone). Protein exhibiting a ratio>2 is indicated with black box. (C) MMP-9 expression in MCF-7 and MDA-MB-468 cells co-cultured with or without (Alone) TAMs for 7 days was assessed by Western blot. β-actin was used as an internal loading control. The blots shown are representative of six independent experiments. (D) MMP-9 expression in MCF-7 and MDA-MB-468 cells co-cultured with or without (Alone) TAMs transfected with adenoviral COX-2 or siRNA COX-2 for 7 days was assessed by Western blot. β-actin was used as an internal loading control. The blots shown are representative of six independent experiments. (E) MMP-9 activity in supernatant collected from different cancer cells was detected using an active human MMP-9 fluorescence assay. The experiments were performed thrice in triplicate. The data are presented as the mean ± SD. **p < 0.01 (versus Alone group).
COX-2 in TAMs induces EMT in breast cancer cells
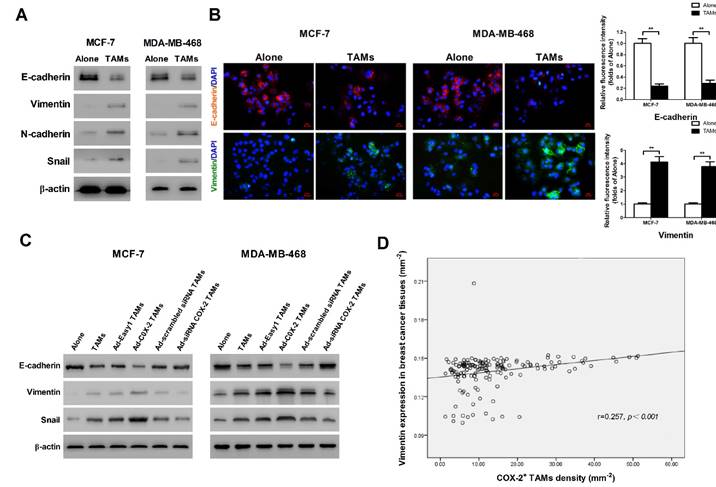
It has been shown that epithelial-mesenchymal transition (EMT) plays a critical role in promoting metastasis in cancer [16]. TAMs were reported to induce EMT in pancreatic cancer or hepatocellular carcinoma through different signal pathways [17]. In order to identify whether COX-2 in TAMs affect the EMT in breast cancer cells, EMT markers were measured by Western blot. We found that TAMs inhibited the expression of epithelial marker E-cadherin and enhanced the expression of mesenchymal markers including Vimentin, N-cadherin and Snail in breast cancer cells (Figure 3A). Indirect immunofluorescence analysis also showed that TAMs reduced E-cadherin expression and increased Vimentin expression in MCF-7 and MDA-MB-468 breast cancer cells (Figure 3B). Ectopic COX-2 expression in TAMs significantly reinforced, while inhibiting COX-2 in TAMs attenuated, this process (Figure 3C). Consistently, based on our breast tissue array, there was also a significant positive correlation between COX-2+ TAMs and the mesenchymal marker Vimentin in breast cancer tissues (Figure 3D).
COX-2 in TAMs enhances TAMs-induced EMT in breast cancer cells. (A) EMT markers in MCF-7 and MDA-MB-468 cells were assessed by Western blot. β-actin was used as an internal loading control. The blots shown are representative of six independent experiments. (B) Immunostaining showed the downregulation of E-cadherin and upregulation of Vimentin in MCF-7 and MDA-MB-468 cells (original magnification: 400×). The nucleus is stained with DAPI (blue), E-cadherin is stained with DyLight 549 (red), and Vimentin is stained with DyLight 488 (green). The experiments were performed thrice in triplicate. The data are presented as the mean ± SD. **p<0.01 (versus Alone group). (C) EMT markers in MCF-7 and MDA-MB-468 cells co-cultured with or without (Alone) TAMs for 7 days were assessed by Western blot. β-actin was used as an internal loading control. The blots shown are representative of six independent experiments. (D) Correlation of COX-2+ TAMs and Vimentin in breast cancer tissues (n = 160) was analyzed by Pearson's correlation analysis.
COX-2 induces PGE2 and IL-6 release from TAMs
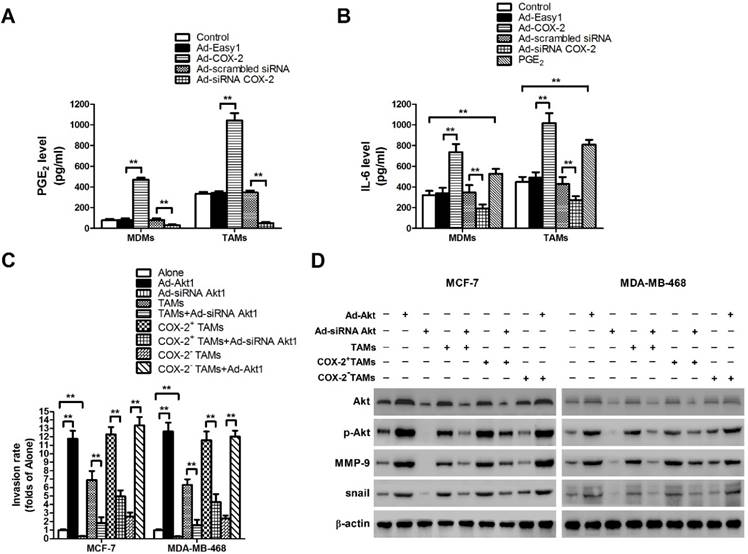
Our previous study showed that COX-2 was essential for the induction and maintenance of M2 polarity in TAMs, which suggested that COX-2 in TAMs might promote cancer progression by inducing various tumor-related cytokines [11]. As the key factor of the COX-2 pathway, the level of secreted PGE2 was first checked (Figure 4A). Since IL-6 is closely involved in EMT and cancer metastasis, the effect of COX-2 on IL-6 release was then detected in our study [18]. We found that COX-2 not only induced the secretion of PGE2, but also enhanced the release of IL-6 in TAMs, while knockdown of COX-2 suppressed the effect (Figure 4B).
COX-2 induces PGE2 and IL-6 release in macrophages and regulates MMP-9 and EMT by activating Akt pathway in breast cancer cells. (A) The level of PGE2 in MDMs or TAMs transfected with adenoviral COX-2 or siRNA COX-2 was detected by EIA. The experiments were performed thrice in triplicate. The data are presented as the mean ± SD. **p < 0.01. (B) The level of IL-6 in MDMs or TAMs transfected with adenoviral COX-2 or siRNA COX-2, or treated with PGE2 (1 μM) was detected by ELISA. The experiments were performed thrice in triplicate. The data are presented as the mean ± SD. **p < 0.01. (C) Inhibiting Akt1 in breast cancer cells attenuated cell invasion induced by COX-2 in TAMs. Breast cancer cells transfected with adenoviral siRNA Akt1 or Akt1 were co-cultured with or without (Alone) TAMs transfected with adenoviral COX-2 or siRNA COX-2. The transwell invasion assay was then carried out as described before. (D) Inhibiting Akt1 resulted in the suppression of MMP-9 and snail in breast cancer cells co-cultured with COX-2+ TAMs. The expression of related proteins in breast cancer cells was detected by Western blot. β-actin was used as an internal loading control, and the blots shown are representative of six independent experiments.
The activation of Akt pathway in breast cancer cells is essential for pro-metastatic effect of COX-2+ TAMs
In our previous study, aberrant activation of the Akt pathway was closely implicated in COX-2+ TAMs-induced breast cancer cell survival [11]. Moreover, it was reported that activation of Akt pathway also enhanced cancer metastasis [19]. Therefore, the role of Akt pathway in COX-2+ TAMs-induced breast cancer metastasis was determined in this study. Blocking the Akt pathway in cancer cells restrained COX-2+ TAMs-induced metastatic potential of breast cancer cells (Figure 4C). Meanwhile, down-regulation of the Akt pathway resulted in the suppression of MMP-9 and EMT transcription factor Snail in breast cancer cells (Figure 4D). Our studies strongly suggest that activation of the Akt pathway is closely involved in COX-2+ TAMs-induced pro-tumor activity in breast cancer cells.
COX-2 in TAMs regulates MMP-9 and EMT in vivo
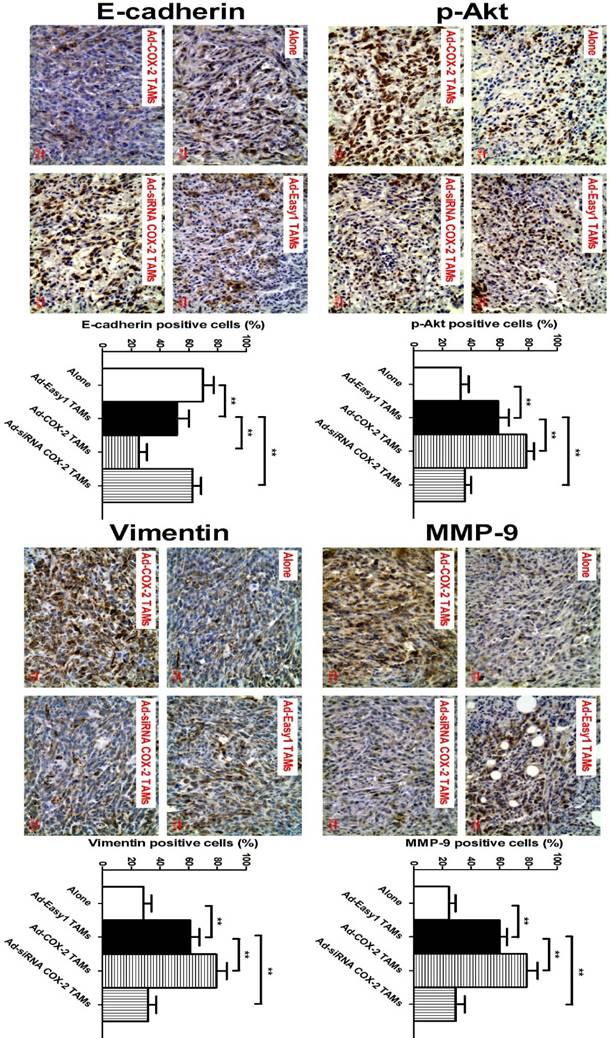
In our previous study, we showed that COX-2 in TAMs promoted breast cancer growth in a mouse xenograft model [11]. Based on this model, we also found that higher expression of MMP-9 and Vimentin, and lower expression of E-cadherin were observed in NOD/SCID mice injected with 4T1 murine breast cancer cells/RAW264.7-derived TAMs, compared with that in mice only injected with 4T1 cells. Consistent with those findings in vitro, significantly increased expression of p-Akt, MMP-9 and Vimentin, and decreased expression of E-cadherin were also detected in the tumor specimens of mice injected with 4T1/COX-2+ TAMs, while an inverse result was obtained from that of mice injected with 4T1/COX-2- TAMs, compared with that of mice injected with 4T1/normal TAMs (Figure 5).
COX-2 in TAMs regulates p-Akt, MMP-9 and EMT in vivo. The animal model was constructed by injecting 4T1 cells alone or with RAW264.7-derived TAMs transfected with adenoviral COX-2 or siRNA COX-2 into the mammary fat pads of NOD/SCID mice. p-Akt, MMP-9, E-cadherin and Vimentin in the tumor tissues were evaluated by IHC (original magnification, ×400). The number of stained and unstained cells was counted to generate the percentage of positive cells in each group. All the data were presented as the mean ± SD, n = 10, *p < 0.05 and **p < 0.01.
Discussion
Metastasis is a major cause of death in patients with malignant tumors. Accumulating studies showed that TAMs promoted the metastatic potential of cancer cells in many kinds of malignant tumors such as lung cancer, papillary thyroid cancer, prostate adenocarcinoma and breast cancer [20-23]. Controlling macrophages infiltration reduced the number of circulating tumor cells and inhibited tumor metastasis in animal models [23,24]. In addition, macrophages in tumor microenvironment are shown to add to the invasive hallmarks of cancer cells by secreting various cytokines (IL-8, CCL5, CXCL8, CXCL16, etc.) [21,23,25,26]. Our previous study showed that COX-2 overexpression in breast TAMs was associated with lymph node metastasis and poor prognosis in breast cancer patients [11]. In this study, we confirmed the contributions of COX-2 in TAMs to the metastatic potential of breast cancer cells. These studies indicate that COX-2 indeed exerts an important effect on the pro-tumor function of TAMs.
As the principle enzymatic product, PGE2 plays a key role in the biological function of COX-2. COX-2/PGE2 enhances cancer invasion/metastasis via activating different downstream signal pathways [27,28]. However, our previous study demonstrated that PGE2 was not the only direct mediator involved in the pro-tumor effect induced by COX-2 in TAMs. COX-2 may also exert pro-tumor effect by causing TAMs to release pro-tumor cytokines through inducing and maintaining M2 macrophage polarity [11]. IL-6 is one of the important cytokines secreted by macrophages, particularly M2 macrophages [25,29]. Accumulating evidence suggests that IL-6 is able to promote cancer cell metastasis [30,31]. Here, we showed that COX-2 enhanced the release of IL-6 from macrophages, which suggested that COX-2 played a multi-faceted pro-tumor effect.
During the metastatic cascades, cancer cells often invade normal tissues through degrading the basement membrane and connective tissues, while MMP/TIMP is closely involved in the process. It was reported that TAMs increased proliferation, migration, and MMP-2 and MMP-9 proteolytic activity in pancreatic cancer cells [32]. In addition, TAMs was also shown to increase invasion of human basal cell carcinoma (BCC) cells by activating COX-2 and release of MMP-9 in BCC cells [33]. In our study, we confirmed that TAMs induced MMP-9 expression and activity in breast cancer cells. More importantly, we clarified that COX-2 in TAMs was an essential factor in regulating MMP-9 in cancer cells. As an important member of the MMPs family, MMP-9 strongly promotes cancer development, invasion and metastasis. MMP-9 is overexpressed in breast cancer and regarded as an effective indicator of breast cancer prognosis [34-37]. Both of PGE2 and IL-6 were reported to enhance MMP-9 level, which may be the reason why COX-2+ TAMs induced higher MMP-9 expression in breast cancer cells [38-41]. In addition to MMPs, EMT is also closely linked to breast cancer metastasis, and can be driven by macrophages in tumor microenvironment [17,42]. In this study, we confirmed the promotion of TAMs on EMT, and further found COX-2 in TAMs had a significant regulatory function on EMT in breast cancer cells. It has been proved that COX-2/PGE2 and IL-6 could induce EMT in human cancers, which strongly suggest that inducing the secretion of pro-metastatic cytokines from macrophages is closely involved in the pro-tumor effect of COX-2 in TAMs [43-45].
Numerous studies show that the Akt pathway is closely involved in cancer metastasis [19,46]. MMP-9 has been considered as a potential downstream signal molecule of the Akt pathway. Some potential anticancer agents such as formononetin and piceatannol were shown to inhibit breast cancer cell invasion by suppressing MMP-9 through blocking the Akt pathway [47,48]. Moreover, the activation of Akt axis is also emerging as a central feature of EMT [49]. Studies including our previous study demonstrated that TAMs induced the activation of Akt pathway in cancer cells, as products secreted from TAMs served as the potential activators of this pathway [11,50,51]. Here, we further identified that the Akt pathway played a critical role in COX-2+ TAMs-induced cancer cell invasion by regulating MMP-9 and EMT process. PGE2 and IL-6 were reported to promote the metastatic potential of cancer cells via Akt pathway [52-55]. In addition, other cytokines such as TGF-β and IL-10, which can be released from M2 macrophages, also have the ability to enhance cancer metastasis by sensitizing the Akt pathway [56,57]. These factors may all contribute to COX-2+ TAMs-mediated cancer cell invasion by modulating the Akt pathway in breast cancer cells.
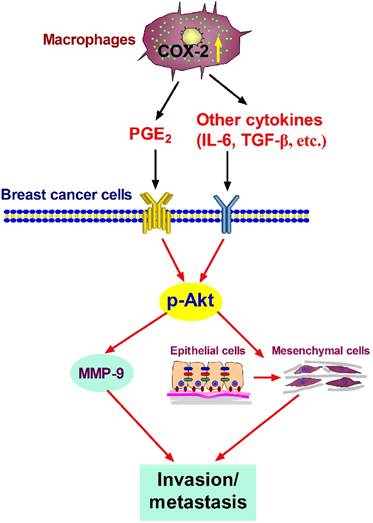
In summary, our studies show that COX-2 in TAMs enhances the metastatic potential of breast cancer cells (Figure 6). As we mainly focused on the effect of COX-2+ TAMs on breast cancer cells, we did not continue investigating the reasons why high COX-2 was expressed in TAMs. In our study, we got TAMs in vitro by co-culturing normal macrophages with malignant breast cancer cells. Therefore, the high expression of COX-2 in TAMs should be induced by the interaction between TAMs and cancer cells in our study. Furthermore, our previous study found that COX-2 in TAMs induced the expression of COX-2 in breast cancer cells, which in turn promoted M2 macrophage polarization [11]. It might be one of the reasons why TAMs exert higher COX-2 expression. In addition, considering that TAMs are one type of stromal cells in the real tumor microenvironment, it is also possible that other components such as other stromal cells and various cytokines have potential effects on COX-2 expression in TAMs. Further investigations focusing on these questions are warranted in our future study. COX-2 is regarded as an essential factor linking inflammation and cancer. Clinical studies have noted that selective or non-selective COX-2 inhibition with COX-2 inhibitors or non-steroidal anti-inflammatory drugs (NSAIDs) have significant chemo-preventive effects on a wide variety of cancers [58,59]. Future studies focusing on COX-2 in the tumor microenvironment will help understand the detailed molecular mechanism involving COX-2 for improving anticancer therapy.
Proposed mechanism showing COX-2+ TAMs-mediated cell metastasis in breast cancer.
Acknowledgements
This study was supported by National Natural Science Foundation of China (NO. 81472475 and NO. 81102007), Health and Family Planning Commission of Chongqing (NO. 2011-2-008), and Scientific Research Foundation of Chongqing Medical University (NO. 201408).
Authors Contributions
Designed the experiments: HZL and GSR. Performed the experiments: LG, JH, ZQ, YHL and HYH. Analyzed the data: LG, JH and TLH. Contributed reagents/materials/analysis tools: TXX, JYW and YL. Manuscript preparation: LG, HZL and GSR.
Competing interest
The authors declare no conflict of interest in this study.
References
1. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49-61
2. Ostuni R, Kratochvill F, Murray PJ. et al. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol. 2015;36:229-39
3. Ramanathan S, Jagannathan N. Tumor associated macrophage: a review on the phenotypes, traits and functions. Iran J Cancer Prev. 2014;7:1-8
4. Sica A, Larghi P, Mancino A. et al. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008;18:349-55
5. Misra S, Sharma K. COX-2 signaling and cancer: new players in old arena. Curr Drug Targets. 2014;15:347-59
6. Khan Z, Khan N, Tiwari RP. et al. Biology of Cox-2: an application in cancer therapeutics. Curr Drug Targets. 2011;12:1082-93
7. Vendramini-Costa DB, Carvalho JE. Molecular link mechanisms between inflammation and cancer. Curr Pharm Des. 2012;18:3831-52
8. Greenhough A, Smartt HJ, Moore AE. et al. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009;30:377-86
9. Bianchini F, Massi D, Marconi C. et al. Expression of cyclo-oxygenase-2 in macrophages associated with cutaneous melanoma at different stages of progression. Prostaglandins Other Lipid Mediat. 2007;83:320-8
10. Tsai CS, Chen FH, Wang CC. et al. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int J Radiat Oncol Biol Phys. 2007;68:499-507
11. Li H, Yang B, Huang J. et al. Cyclooxygenase-2 in tumor-associated macrophages promotes breast cancer cell survival by triggering a positive-feedback loop between macrophages and cancer cells. Oncotarget. 2015;6:29637-50
12. Zheng Y, Cai Z, Wang S. et al. Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug-induced apoptosis. Blood. 2009;114:3625-8
13. Li H, Huang J, Yang B. et al. Mangiferin exerts antitumor activity in breast cancer cells by regulating matrix metalloproteinases, epithelial to mesenchymal transition, and beta-catenin signaling pathway. Toxicol Appl Pharmacol. 2013;272:180-90
14. Luo YP, Zhou H, Krueger J. et al. The role of proto-oncogene Fra-1 in remodeling the tumor microenvironment in support of breast tumor cell invasion and progression. Oncogene. 2010;29:662-73
15. Rydlova M, Holubec L Jr, Ludvikova M Jr. et al. Biological activity and clinical implications of the matrix metalloproteinases. Anticancer Res. 2008;28:1389-97
16. Gomes LR, Terra LF, Sogayar MC. et al. Epithelial-mesenchymal transition: implications in cancer progression and metastasis. Curr Pharm Biotechnol. 2011;12:1881-90
17. Zhang J, Yao H, Song G. et al. Regulation of epithelial-mesenchymal transition by tumor-associated macrophages in cancer. Am J Transl Res. 2015;7:1699-711
18. Ho LJ, Luo SF, Lai JH. Biological effects of interleukin-6: Clinical applications in autoimmune diseases and cancers. Biochem Pharmacol. 2015;97:16-26
19. Polivka J Jr, Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther. 2014;142:164-75
20. Trikha P, Sharma N, Pena C. et al. E2f3 in tumor macrophages promotes lung metastasis. Oncogene. 2015
21. Fang W, Ye L, Shen L. et al. Tumor-associated macrophages promote the metastatic potential of thyroid papillary cancer by releasing CXCL8. Carcinogenesis. 2014;35:1780-7
22. Hu W, Qian Y, Yu F. et al. Alternatively activated macrophages are associated with metastasis and poor prognosis in prostate adenocarcinoma. Oncol Lett. 2015;10:1390-6
23. Datar I, Qiu X, Ma HZ. et al. RKIP regulates CCL5 expression to inhibit breast cancer invasion and metastasis by controlling macrophage infiltration. Oncotarget. 2015;6:39050-61
24. Liu C, Li Z, Wang L. et al. Activating transcription factor 4 promotes angiogenesis of breast cancer through enhanced macrophage recruitment. Biomed Res Int. 2015;2015:974615
25. Xu H, Lai W, Zhang Y. et al. Tumor-associated macrophage-derived IL-6 and IL-8 enhance invasive activity of LoVo cells induced by PRL-3 in a KCNN4 channel-dependent manner. BMC Cancer. 2014;14:330
26. Cho SW, Kim YA, Sun HJ. et al. CXCL16 signaling mediated macrophage effects on tumor invasion of papillary thyroid carcinoma. Endocr Relat Cancer. 2015
27. Kim MJ, Kim HS, Lee SH. et al. NDRG2 controls COX-2/PGE(2)-mediated breast cancer cell migration and invasion. Mol Cells. 2014;37:759-65
28. Reader J, Holt D, Fulton A. Prostaglandin E2 EP receptors as therapeutic targets in breast cancer. Cancer Metastasis Rev. 2011;30:449-63
29. Heusinkveld M, de Vos van Steenwijk PJ, Goedemans R. et al. M2 macrophages induced by prostaglandin E2 and IL-6 from cervical carcinoma are switched to activated M1 macrophages by CD4+ Th1 cells. J Immunol. 2011;187:1157-65
30. Tawara K, Oxford JT, Jorcyk CL. Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: potential of anti-IL-6 therapies. Cancer Manag Res. 2011;3:177-89
31. Walter M, Liang S, Ghosh S. et al. Interleukin 6 secreted from adipose stromal cells promotes migration and invasion of breast cancer cells. Oncogene. 2009;28:2745-55
32. Liu CY, Xu JY, Shi XY. et al. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab Invest. 2013;93:844-54
33. Tjiu JW, Chen JS, Shun CT. et al. Tumor-associated macrophage-induced invasion and angiogenesis of human basal cell carcinoma cells by cyclooxygenase-2 induction. J Invest Dermatol. 2009;129:1016-25
34. Mehner C, Hockla A, Miller E. et al. Tumor cell-produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple negative breast cancer. Oncotarget. 2014;5:2736-49
35. Gao J, Liu X, Yang F. et al. By inhibiting Ras/Raf/ERK and MMP-9, knockdown of EpCAM inhibits breast cancer cell growth and metastasis. Oncotarget. 2015;6:27187-98
36. Bottino J, Gelaleti GB, Maschio LB. et al. Immunoexpression of ROCK-1 and MMP-9 as prognostic markers in breast cancer. Acta Histochem. 2014;116:1367-73
37. Yousef EM, Tahir MR, St-Pierre Y. et al. MMP-9 expression varies according to molecular subtypes of breast cancer. BMC Cancer. 2014;14:609
38. Yen JH, Kocieda VP, Jing H. et al. Prostaglandin E2 induces matrix metalloproteinase 9 expression in dendritic cells through two independent signaling pathways leading to activator protein 1 (AP-1) activation. J Biol Chem. 2011;286:38913-23
39. Itatsu K, Sasaki M, Yamaguchi J. et al. Cyclooxygenase-2 is involved in the up-regulation of matrix metalloproteinase-9 in cholangiocarcinoma induced by tumor necrosis factor-alpha. Am J Pathol. 2009;174:829-41
40. Zergoun AA, Zebboudj A, Sellam SL. et al. IL-6/NOS2 inflammatory signals regulate MMP-9 and MMP-2 activity and disease outcome in nasopharyngeal carcinoma patients. Tumour Biol. 2015
41. Sun W, Liu DB, Li WW. et al. Interleukin-6 promotes the migration and invasion of nasopharyngeal carcinoma cell lines and upregulates the expression of MMP-2 and MMP-9. Int J Oncol. 2014;44:1551-60
42. Wang Y, Zhou BP. Epithelial-mesenchymal Transition--A Hallmark of Breast Cancer Metastasis. Cancer Hallm. 2013;1:38-49
43. Qin G, Xu F, Qin T. et al. Palbociclib inhibits epithelial-mesenchymal transition and metastasis in breast cancer via c-Jun/COX-2 signaling pathway. Oncotarget. 2015
44. Bocca C, Ievolella M, Autelli R. et al. Expression of Cox-2 in human breast cancer cells as a critical determinant of epithelial-to-mesenchymal transition and invasiveness. Expert Opin Ther Targets. 2014;18:121-35
45. Lee SO, Yang X, Duan S. et al. IL-6 promotes growth and epithelial-mesenchymal transition of CD133+ cells of non-small cell lung cancer. Oncotarget. 2015
46. Wu X, Sun L, Wang X. et al. Breast Cancer Invasion and Metastasis by mPRalpha Through the PI3K/Akt Signaling Pathway. Pathol Oncol Res. 2015
47. Zhou R, Xu L, Ye M. et al. Formononetin inhibits migration and invasion of MDA-MB-231 and 4T1 breast cancer cells by suppressing MMP-2 and MMP-9 through PI3K/AKT signaling pathways. Horm Metab Res. 2014;46:753-60
48. Ko HS, Lee HJ, Kim SH. et al. Piceatannol suppresses breast cancer cell invasion through the inhibition of MMP-9: involvement of PI3K/AKT and NF-kappaB pathways. J Agric Food Chem. 2012;60:4083-9
49. Larue L, Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3' kinase/AKT pathways. Oncogene. 2005;24:7443-54
50. Chen J, Yao Y, Gong C. et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell. 2011;19:541-55
51. Li GQ, Xie J, Lei XY. et al. Macrophage migration inhibitory factor regulates proliferation of gastric cancer cells via the PI3K/Akt pathway. World J Gastroenterol. 2009;15:5541-8
52. Ho MY, Hung SW, Liang CM. et al. Recombinant viral capsid protein VP1 suppresses lung cancer metastasis by inhibiting COX-2/PGE2 and MIG-7. Oncotarget. 2014;5:3931-43
53. Du M, Shi F, Zhang H. et al. Prostaglandin E2 promotes human cholangiocarcinoma cell proliferation, migration and invasion through the upregulation of beta-catenin expression via EP3-4 receptor. Oncol Rep. 2015;34:715-26
54. Jee SH, Chu CY, Chiu HC. et al. Interleukin-6 induced basic fibroblast growth factor-dependent angiogenesis in basal cell carcinoma cell line via JAK/STAT3 and PI3-kinase/Akt pathways. J Invest Dermatol. 2004;123:1169-75
55. Kobayashi S, Werneburg NW, Bronk SF. et al. Interleukin-6 contributes to Mcl-1 up-regulation and TRAIL resistance via an Akt-signaling pathway in cholangiocarcinoma cells. Gastroenterology. 2005;128:2054-65
56. Li Y, Jia L, Liu C. et al. Axl as a downstream effector of TGF-beta1 via PI3K/Akt-PAK1 signaling pathway promotes tumor invasion and chemoresistance in breast carcinoma. Tumour Biol. 2015;36:1115-27
57. Sung WW, Lee H. The role of interleukin-10 in the progression of human papillomavirus-associated lung carcinoma. Oncoimmunology. 2013;2:e25854
58. Patrignani P, Patrono C. Cyclooxygenase inhibitors: From pharmacology to clinical read-outs. Biochim Biophys Acta. 2015;1851:422-32
59. Ghosh N, Chaki R, Mandal V. et al. COX-2 as a target for cancer chemotherapy. Pharmacol Rep. 2010;62:233-44
Author contact
![]() Corresponding authors: Hongzhong Li, E-mail: tibetlhzcom. Or Guosheng Ren, E-mail: rengs726com. Chongqing Key Laboratory of Molecular Oncology and Epigenetics, The First Affiliated Hospital of Chongqing Medical University, No.1 Youyi Road, Yuzhong District, Chongqing, 400016, China. Phone: 0086-2389011477; Fax: 0086-2389012305.
Corresponding authors: Hongzhong Li, E-mail: tibetlhzcom. Or Guosheng Ren, E-mail: rengs726com. Chongqing Key Laboratory of Molecular Oncology and Epigenetics, The First Affiliated Hospital of Chongqing Medical University, No.1 Youyi Road, Yuzhong District, Chongqing, 400016, China. Phone: 0086-2389011477; Fax: 0086-2389012305.