Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(12):1520-1531. doi:10.7150/ijbs.21107 This issue Cite
Research Paper
Dl-3-n-butylphthalide prevents the disruption of blood-spinal cord barrier via inhibiting endoplasmic reticulum stress following spinal cord injury
Binbin Zheng1,2,3*, Yulong Zhou2,3*, Hongyu Zhang3, Guangyong Yang1, Zhenghua Hong1, Dandan Han1, Qingqing Wang2, Zili He2, Yanlong Liu3, Fenzan Wu4, Xie Zhang5, Songlin Tong4, Huazi Xu2 ![]() , Jian Xiao3
, Jian Xiao3 ![]()
1. Department of Orthopaedics, Taizhou Hospital, Wenzhou Medical University, Linhai, Zhejiang, 317000 PR China;
2. Key Laboratory of Orthopaedics of Zhejiang Province, Department of Orthopaedics, The Second Affiliated Hospital, Wenzhou Medical University, Wenzhou, Zhejiang, 325035 PR China;
3. Molecular Pharmacology Research Center, School of Pharmacy, Wenzhou Medical University, Wenzhou, Zhejiang, 325035 PR China;
4. Department of Neurosurgery, Affiliated Cixi People's Hospital, Wenzhou Medical University, Ningbo, Zhejiang, 315300, PR China;
5. Department of Gastroenterology, Ningbo Medical Treatment Center Li Hui-li Hospital, Ningbo, Zhejiang, 315040, PR China.
* These authors had equal contribution and are designated as co-first authors.
Received 2017-5-20; Accepted 2017-10-15; Published 2017-11-27
Abstract

After spinal cord injury (SCI), the destruction of blood-spinal cord barrier (BSCB) is shown to accelerate gathering of noxious blood-derived components in the nervous system, leading to secondary neurodegenerative damages. SCI activates endoplasmic reticulum stress (ER stress), which is considered to evoke secondary damages of neurons and glia. Recent evidence indicates that Dl-3-n-butylphthalide (NBP) has the neuroprotective effect in ischaemic brain injury, but whether it has protective effects on SCI or not is largely unclear. Here, we show that NBP prevented BSCB disruption after SCI via inhibition of ER stress. Following a moderate contusion injury of the T9 level of spinal cord, NBP was administered by oral gavage and further treated once a day. NBP significantly attenuated BSCB permeability and breakdown of adherens junction (AJ) and tight junction (TJ) proteins, then improved locomotion recovery following SCI. The protective role of NBP on BSCB disruption is associated with the restrain of ER stress caused by SCI. Furthermore, NBP considerably constrained the expression of ER stress-associated proteins and degradation of TJ and AJ in human brain microvascular endothelial cells (HBMECs) treated with TG. In conclusion, our results indicate that ER stress is associated with the disruption of BSCB integrity after injury, NBP attenuates BSCB disruption via inhibiting ER stress and improve functional recovery following SCI.
Keywords: Dl-3-n-butylphthalide, Blood-spinal cord barrier, Spinal cord injury, Endocytoplasmic reticulum stress.
Introduction
As a highly specialized spinal cord endothelial structure, the blood spinal cord barrier (BSCB) is the functional equivalent of the blood brain barrier (BBB) and provides a particular mini-environment of the spinal cord for the cellular constituents [1]. Impairment of the BSCB plays an important role in the pathogenesis or development of some pathological factors of spinal cord, just like SCI and amyotrophic lateral sclerosis. When the BSCB integrity is damaged after spinal cord injury (SCI), blood cells and plasma components cross into the spinal cord parenchyma, contributing to secondary damages, such as inflammation, local edema, focal hemorrhage and free radical stress [2, 3]. These secondary injuries cause the ''programmed death'' of glia and neurons, generating perpetual neurological deficiency [4, 5]. Therefore, drugs that targeting the disruption of BSCB may facilitate the restriction of cellular damage and functional recovery following SCI.
The endoplasmic reticulum (ER) is a vital subcellular organoid for the synthesis and folding of secreted and membrane-bound proteins in eukaryotic cells [6, 7], which are essential for natural cellular function and the survival of cell. ER stress can be triggered by various exogenous stressors, such as exhaustion of ER Ca2+ stores, collection of misfolded or unfolded proteins [8-10]. It plays a vital role in a plenty of neural diseases, such as neurodegenation disorders, sclerosis, SCI, and cerebral ischemia [11-13]. Previous reports show that ER stress is activated in spinal cord after SCI, among which oligodendrocytes and neurons are most investigated [14, 15]. During SCI, excessive ER stress that results in neural apoptosis is associated with the GRP78 protein, the transcription stimulation of CHOP and the triggering of ER-related caspase-12 [16]. Although ER stress is proved plays a vital role in SCI, the effect of ER stress on the BSCB disruption after SCI is little known. Elucidating the impact of ER stress on the disruption of BSCB and the adjustment of the ER stress-associated path may present an underlying point for SCI-induced BSCB dysfunction and improve functional recovery of SCI. Furthermore, NBP shows neuroprotective effects through various mechanisms, but the influence of NBP on ER stress following SCI remains unclear.
DL-3-n-butylphthalide (NBP), initially extracted from seeds of Apium graveolens Linn, Chinese celery, is a new and powerful natural free radical scavenger [17]. Previous studies demonstrate that NBP has neuroprotective effects by acting through multiple-targets. The underlying molecular mechanisms may include accelerating vasculogenesis via up-regulation of HIF-1alpha, vascular endothelial growth factor expression and improving microcirculation dysfunction during ischaemia [18], inhibition of the inflammatory response [19], decreasing of oxidative impairment [17, 20], amelioration of mitochondrial function, decreasing of neuronal apoptosis [18, 21]. Meanwhile, NBP also reduce blood brain barrier (BBB) damage in cerebral ischaemia model [22, 23]. However, little is known that the effect of NBP on the BSCB subjected to SCI. In our study, we investigated the function of NBP on BSCB disruption and the involvement of ER stress in the disruption of BSCB following SCI.
In the present study, we demonstrate that ER stress is related to the BSCB disruption induced by SCI. NBP prevents the degradation of adherens junction (AJ) and tight junction (TJ) protein, attenuates BSCB permeability via inhibiting ER stress and improves functional recovery following SCI. Our results indicate an underlying target for SCI treatment and drug development.
Materials and Methods
Reagents and antibodies
NBP, a yellow oil-like liquid with a purity of 98.6%, was got from (Shijiazhuang Pharmaceutical Group Enbipu Pharmaceutical Co., Ltd, Shijiazhuang, Hebei, China). Human brain microvascular endothelial cell (HBMEC) was bought from ScienCell Research Laboratories (San Diego, CA, USA). Texas red-conjugated anti-IgG and Antibodies against GRP78, Occludin, CHOP, Claudin5, CD31, GAPDH were bought from Santa (Santa Cruz Biotechnology, CA, USA). Alexa Fluor 488-conjugated anti-IgG and Anti-p120-Catenin, XBP-1, β-Catenin, ATF6, Cleaved caspase-12, ATF4, and PDI were bought from Abcam (Abcam, Cambridge, Britain).
SCI model
The adult female Sprague-Dawley rats (220 - 250 g) were obtained from the Animal Center of the Chinese Academy of Sciences in Shanghai, China. The care and use of animals was conformed to the Guide from the National Institutes of Health. All the rats were housed under standard temperature conditions. Rats were randomly divided into the Sham, SCI, and NBP treatment groups. The animals were anaesthetized with 10 % chloralic hydras (3.5 mL/kg, i.p.) and a laminectomy was performed at the T9 level. The moderate contusion injury was performed on exposed spinal cord with a vascular clip (30 g forces, Oscar, China) for 2 minutes. Bilateral cord hematoma and immediate postoperative paraplegia were used as inclusion criteria. The mortality rate of the SCI model is 1%. NBP, dissolved in vegetable oil at the concentration of 80 mg/ml, was administered by oral gavage (80 mg/kg) after injury [24] and treated once a day for indicated time points of experiment. The rats of Sham group just performed a T9 laminectomy without spinal cord injury and received vehicle solution. Postoperative monitoring involved manual bladder emptying twice a day and the administration of cefazolin sodium (50 mg/kg, i.p.).
Cell culture
HBMECs were cultured in Endothelial Cell Medium and maintained in a 37 °C atmosphere containing 5 % CO2. NBP involved dilution to a stockpile liquid of 30 μM in DMSO. NBP (30 μM), Thapsigargin (TG, 10 μM) compound with NBP (30 μM) and TG were separately treated in the cells. All experiments were achieved in triplicate.
Behavioral tests
After SCI, Functional deficit was examined with The Basso, Beattie and Bresnahan (BBB) scores as previously described [25]. The BBB score was evaluated by two blinded independent inspectors. It was evaluated at 7 days post-operation in an open field scale by two blinded independent inspectors. In a few words, the BBB scores range from 0 score (no limb movement or weight support) to 21 scores (normal locomotion). It was acted on the basis of the normal progress of movement recovery in thoracic SCI rats.
Evans blue dye assays
The BSCB permeability was observed with Evans blue dye extravasation as described previously [26]. 2 % Evans blue dye (EB, Sigma-Aldrich, St. Louis, MO, USA, 2 ml/kg) was injected in rats via the tail vein at 24 h post-operation. Two hours later, some rats were anesthetized and sacrificed. The T9 spinal cord tissues were immersed in N, N'-dimethylformamide (DMF, JinSan, Wenzhou, China) for 72 h at 50 °C. The fluorescence of the supernatant liquid was quantified using enzyme-labelled meter at an excitation wavelength of 620 nm and an emission wavelength of 680 nm. In addition, some rats were anesthetized and perfused with 4 % paraformaldehyde. The spinal cord tissues were sectioned into 20 μm slices by using cryostat, the fluorescence was observed.
Paracellular Permeability Assay
HBMECs were cultured at a density of 1×105 cells/well in PET membrane 24-well cell culture inserts with 0.4 µm pore size overnight (Corning Life Sciences, Corelle, New York, USA), then TG, TG compound with NBP was treated for 6 h, next FITC-dextran (1 mg/mL, Sigma-Aldrich) was put in cell culture for another 2 h. Last, the fluorescence of FITC-dextran infiltrated into the lower chambers examined with enzyme-labelled meter at an excitation wavelength of 493 nm and an emission wavelength of 517 nm.
Western blot analysis
Protein from animals or HBMECs was distilled with protein extraction reagents. 50 μg proteins was divided with 12 % gel, then diverted onto a PVDF membrane (Bio-Rad, Hercules, CA, USA). The membrane was blockaded with 5 % milk in TBST for 60 minutes, and incubated at 4 °C overnight with primary antibody solutions: Occludin (1:800), Claudin-5 (1:800), PDI (1:1000), β-Catenin (1:1000), CHOP (1:300), GRP78 (1:300), p120--Catenin (1:1000), XBP-1 (1:1000), ATF4 (1:1000), ATF6 (1:1000), GAPDH (1:300) and Cleaved-caspase12 (1:1000). TBS was used to wash the membranes 5 minutes for three times, then the membranes were incubated secondary antibodies at room temperature for 2 h. Images were visualized by the ChemiDicTM XRS + Imaging System (Bio-Rad). All experiments were repeated in triplicate.
Immunofluorescence staining
T9 spinal cord tissues got from rats at 24 h post-SCI were fixed for 6 h in 4% paraformaldehyde, inserted in paraffin and cuted into 5 μm section. The slices were hatched with 5 % BSA for 60 minutes at room temperature and hatched with primary antibodies overnight at 4 °C: Claudin-5 (1:100), Chop (1:100), p120-Catenin (1:200) and CD31 (1:100). The slices were washed with PBS 5 minutes for three times and then incubated with Texas red-conjugated anti-IgG (1:100) or Alexa Fluor 488-conjugated anti-IgG (1:500). The nuclei was coloured by Hoechst 33258 (0.25 lg/mL, Beyotime Institute of Biotechnology, Shanghai, China). HBMECs, which grew on microscopic glass of 14×14 mm, were washed with PBS, fixed for 30 min by 4 % paraformaldehyde, then washed with PBS and blockaded for 60 min with 5 % bovine serum albumin (BSA). Then cells were hatched with primary antibodies overnight at 4 °C (p120-Catennin (1:200), Claudin-5 (1:50), PDI (1:200)). Cells were washed with PBS 5 minutes for three times and then incubated with Texas red-conjugated anti-IgG (1:100) or Alexa Fluor 488-conjugated anti-IgG (1:500) at room temperature for 60 min. The nuclei was coloured by Hoechst 33258. Images were visualized with Nikon A1 plus confocal microscope (Nikon, Japan).
Statistical analysis
Data are presented as the Mean ± SEM. When there were two experimental groups, statistical significance was tested with Student's t-test. When there were three or more experimental groups, statistical significance was checked with one-way analysis of variance (ANOVA) and Tukey's multiple comparison post hoc tests. When P values < 0.05, differences of data were thought to be statistically significant.
Results
NBP attenuates BSCB disruption and accelerates locomotion recovery following SCI
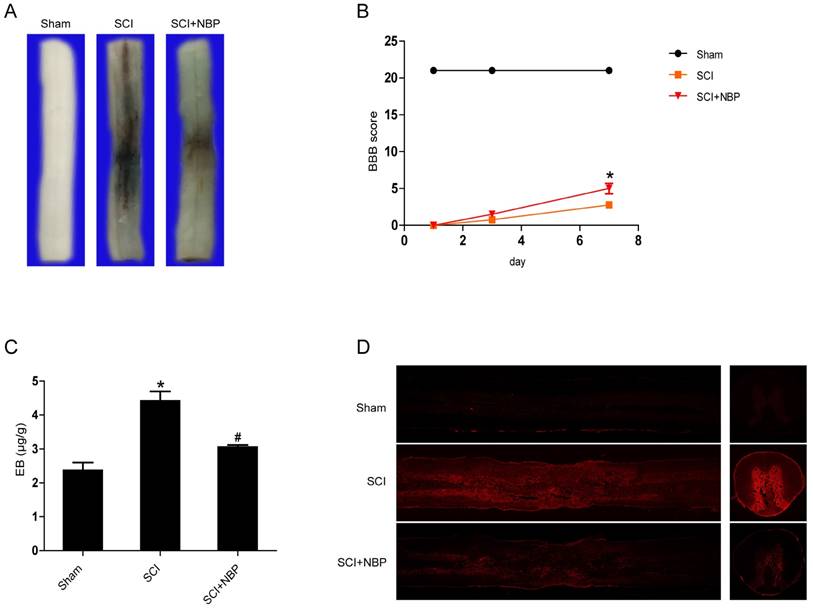
To clarify whether NBP inhibits the increase of BSCB permeability caused by SCI, Evan's Blue assay was examined. We investigated the influence of NBP on the integrity of BSCB at 24 h post-SCI. As the result shown in Figure 1A and C, we found that there was more Evan's Blue dye extravasation in the SCI group than in the Sham group, indicating that SCI elicits BSCB disruption. After treated with NBP, the amount of Evan's Blue dye extravasation in spinal cord tissues was obviously less than in the SCI group. In addition, the fluorescence density of EB in the SCI group was higher than in the Sham group, NBP treatment considerably decreased the fluorescence density (Figure. 1D). Locomotion recovery was assessed in 1 day, 3 day3, and 7 days post-SCI with BBB scores. Compared with the SCI group, NBP management obviously improved the locomotion activity at 7 days post-SCI (Figure. 1B). These data imply that NBP attenuates the disruption of BSCB and improves locomotion recovery following injury.
NBP inhibits BSCB disruption and improves locomotion recovery following SCI. NBP was administered by oral gavage after SCI and the permeability of BSCB was tested at 24 h after injury with Evan's Blue dye. (A) Typical spinal cords dispaly that Evan's Blue dye permeated into the injury spinal cord tissue at 24 h post-SCI. Compared to the SCI group, NBP treatment reduced the quantity of Evan's Blue dye in the surface of spinal cord (n=5 per group). (B) The BBB scores of the Sham, SCI and NBP treatment groups (n=8 per group). NBP improved functional recovery at 7 day after injury. * P < 0.05 versus the SCI group. (C) Quantify of the quantity of Evan's Blue dye of the Sham, SCI and NBP treatment groups at 24 h post-SCI (n=5 per group). # P<0.05 versus the SCI group, * P<0.01 versus the Sham group. (D) Typical confocal pictures of Evan's Blue leakage in each group (n=5 per group).
NBP treatment inhibits the loss of AJ and TJ in SCI model rats
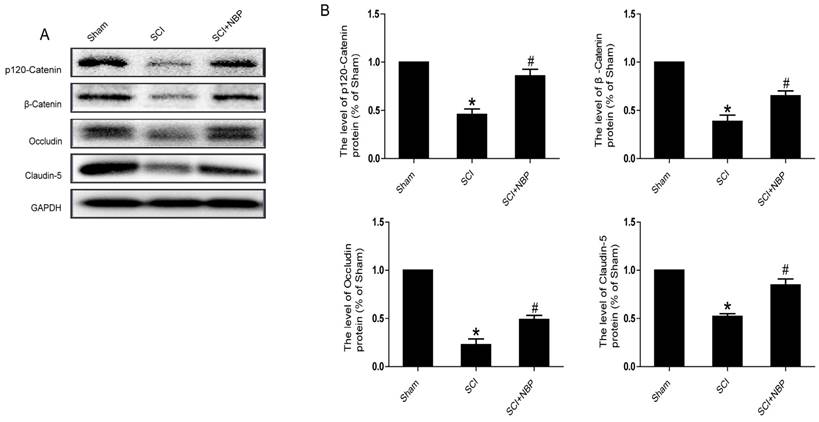
It is known to all that tight junction and adherens junction are involved in BSCB integrity [27]. To verify whether NBP attenuates the increasing of BSCB penetrability by inhibiting the missing of AJ and TJ after injury, we detected the change of SCI-induced AJ and TJ proteins and the effect of NBP on these alterations at 24 h after SCI. As shown in Fig. 2A, the protein expression of TJ (Claudin-5, Occludin) and AJ (β-Catenin, p120-Catenin) was reduced at 24 h post-injury. Furthermore, compared with the SCI group, NBP drastically attenuated the reduction of AJ and TJ proteins expression at 24 h post-SCI (Fig. 2B). In Fig. 4A and B, the fluorescence density of Claudin-5, p120-Catenin and CD31 was reduced in the SCI group and NBP adminstration attenuated the reduction of fluorescence density. The data shown above suggest that NBP attenuates the disruption of BSCB by preventing the depletion of AJ and TJ proteins following injury.
NBP attenuates the breakdown of AJ and TJ proteins following injury. (A) Proteins expression of β-Catenin, p120-Catenin, Claudin-5 and Occludin at the contusion epicenter of spinal cord. At 24 h following injury, the level of AJ and TJ proteins decreased and NBP treatment reversed these changes. (B) Quantify of western blot. # P < 0.05 versus the SCI group, * P<0.01 versus the Sham group (n=5 per group).
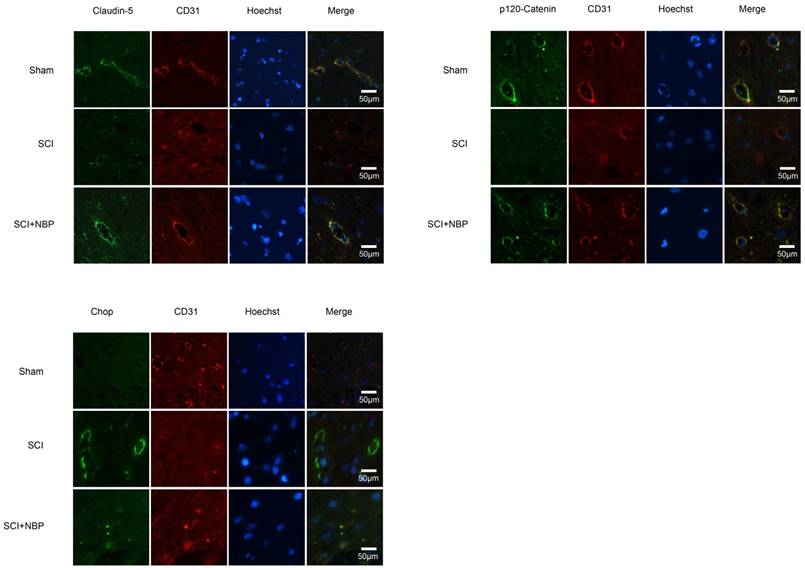
NBP prevents the degradation of p120-Catenin and Claudin-5 and the expression of CHOP in vivo. (A, B) Immunofluorescence displays that Claudin-5 (green) and p120-Catenin (green) co-localized with CD31 (red)-marked blood vessels of the Sham, SCI and NBP treatment groups (n=5 per group). NBP significantly prevented the disruption of Claudin-5 and p120-Catenin compared with SCI group. (C) Immunofluorescence staining results of CHOP (green) colocalize with CD31 (red) -marked blood vessels of spinal cord tissue, nuclei is marked by Hoechst (blue). The level of CHOP obviously reduced after treating NBP (n=5 per group).
NBP suppresses the activation of ER stress after SCI
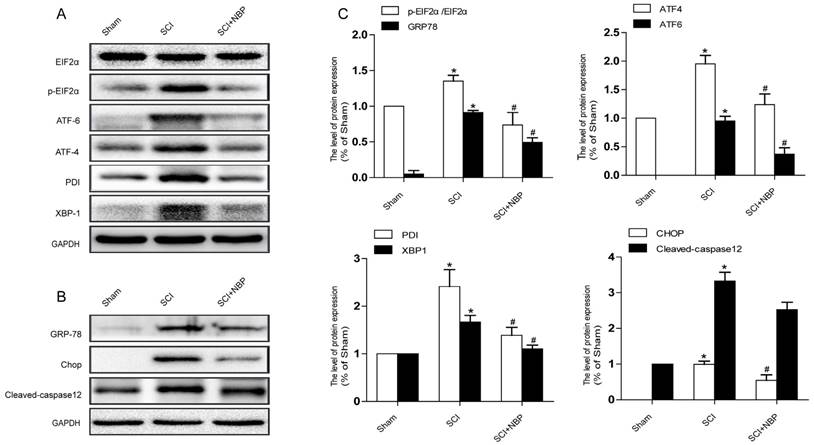
Previous study proves that the level of ER stress is elevated in the spinal cord tissue after SCI [28, 29]. To evaluate the dysfunction of ER after SCI and the effect of NBP on ER stress, we examined ER stress associated proteins including p-EIF 2α/EIF 2α, ATF-4, ATF-6, XBP-1, PDI, GRP78 and CHOP. As shown in Fig.3A, B, the protein expression of EIF 2α/p- EIF 2α, ATF-4, ATF-6, CHOP, XBP-1, PDI and GRP78 increased at 24h post-SCI. Moreover, NBP significantly attenuated the expression of ER stress related proteins at 24h post-SCI compared with SCI group. Immunofluorescence result of CHOP and CD31 is coherent with corresponding results of the western blot (Figure. 4C). The expression of Cleaved-caspase 12 (38 kDa, active form) which is a downstream molecule of CHOP was drastically raised in the SCI group and this increase was markedly alleviated by NBP (Figure. 3A, B and C). The results above show that NBP effectively suppresses the expression of ER stress-related proteins in the injury spinal cord.
NBP inhibits the triggering of ER Stress caused by SCI. (A, B) Typical western blots of ER stress related proteins p-EIF2α/EIF2α, ATF-6, ATF-4, PDI, GRP78, XBP-1, Cleaved-caspase12 and CHOP in the Sham, SCI and SCI treated NBP groups. ER stress was activated at 24 h after SCI and suppressed by treating NBP. (C) Quantify of western blot. # P < 0.05 versus the SCI group, * P < 0.05 versus the Sham group (n=5 per group).
NBP alleviates the decreases of TJ and AJ proteins induced by TG in vitro
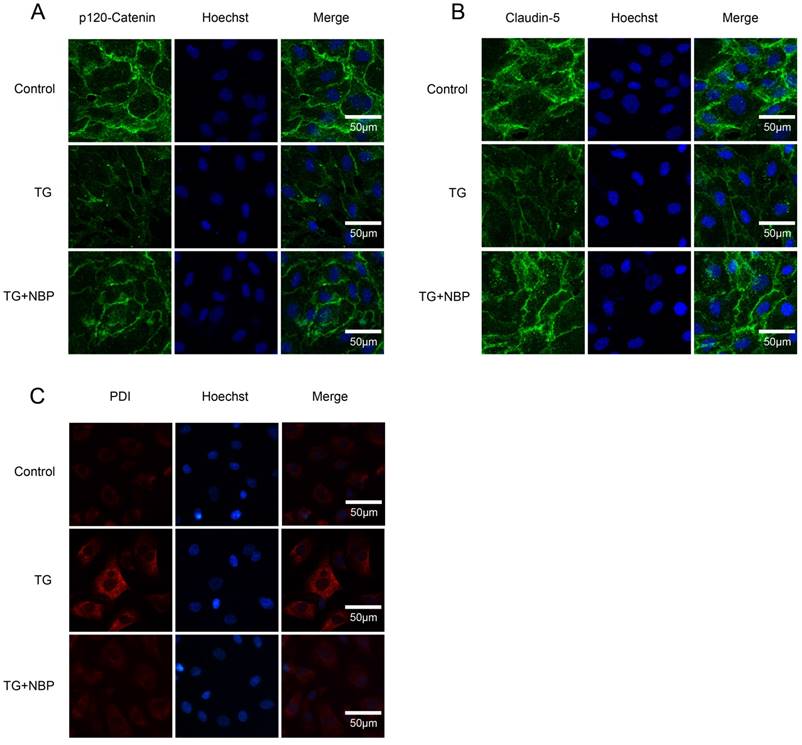
To study the influence of NBP on BSCB integrity in vitro, HBMECs were treated with thapsigargin (TG). We tested the variation of AJ and TJ. The expression of AJ (p120-Catenin, β-Catenin) and TJ (Occludin, Claudin-5) were reduced in the TG group and NBP treatment significantly attenuated these decreasing (Fig. 5A, B and C). In addition, paracellular permeability assessment of FITC-dextran was examined to estimate permeability change of BSCB. Compared with the TG group, the permeability of FITC-dextran was decreased in TG + NBP group (Fig. 5D). Cell immunofluorescence also presented that the fluorescence density of p120-Catenin and Claudin-5 reduced in the TG group and NBP administration attenuated the reduction of corresponding fluorescence intensity (Figure. 7A, B). The data shown above indicate that the protective effect of NBP on the permeability is due to the decrease of TJ and AJ loss after TG administration.
NBP prevents depletion of AJ and TJ proteins in TG-treated cells. (A) Typical western blots of p120-Catenin, β-Catenin, Claudin-5, Occludin in HBMECs. TG reduced the level of AJ and TJ proteins, which was inhibited by NBP. (B, C) Quantification data of β-Catenin, p120-Catenin, Claudin-5, Occludin in each group cells. # P<0.05 versus the TG group, * P<0.05 versus the Control group. (D) Evaluation of paracellular permeability by FITC-dextran in HBMECs. NBP treatment attenuated the disruption of paracellular permeability caused by TG. # P<0.05 versus the TG group, * P<0.01 versus the Control group. All experiments were performed in triplicate.
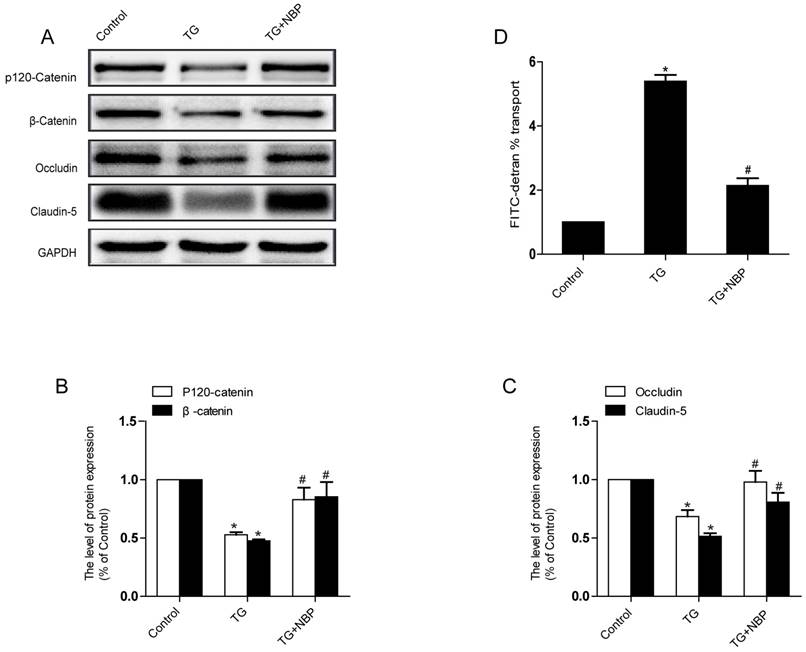
NBP inhibits ER stress caused by TG in vitro
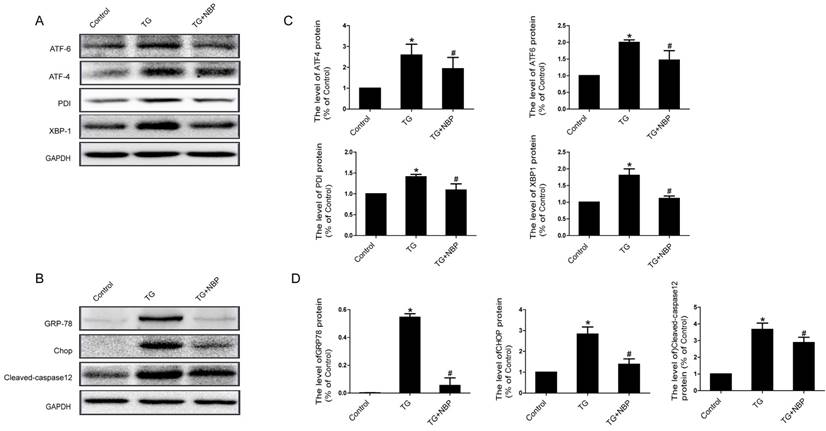
We next explored the influence of NBP on ER stress related proteins. Western blot results presented that the expression of ATF-4, cleaved-caspase 12, XBP-1, CHOP, GRP78, PDI and ATF-6 was considerably increased, and NBP significantly suppressed ER stress related proteins expression (Figure. 6A, B, C and D). Additionally, the result of immunofluorescence intensity of PDI is consistent with that of western blot (Figure. 7C). These results imply that NBP attenuates BSCB disruption via inhibiting ER stress in vitro.
Discussion
Under physiological conditions, an intact BSCB is critically important for building and maintaining a microenvironment that keeps the natural function of axons, neurons and glial cells in spinal cord tissue [1]. Disorder of BSCB function and structure underneath numerous pathological diseases such as stroke and SCI will badly disturb the internal environment in the spinal cord [30, 31]. BSCB disruption has been suggested to accelerate the death of neuronal cells both in traumatic SCI and neurodegenerative diseases [32, 33]. NBP, a neuroprotective drug that is make use of stroke treatment in China, has obviously anti-ischaemic result in the central neural system via diminishing the cerebral infarct zone and improving brain edema in the rates of middle cerebral artery occlusion [34], inhibiting of ischemia-induced oxidative damage and neuron apoptosis after focal cerebral ischemia in mice [35]. Besides, NBP is found to have an effect on ameliorating microcirculation dysfunction and defending endothelial cell against oxidative stress [20, 35]. NBP attenuates BBB disruption and reduced cerebral edema by inhibiting the protein expression of RhoA in cerebral cortex near focal cerebral infarction [36]. Mechanistic studies indicate that NBP leads to less dephosphorylation of Akt and ERK in ischaemic brain, which related to decreased BBB damage as validated by low levels of spectrin breakdown products and matrixmetalloproteinases [22]. Therefore, we postulated that allothogenic NBP may present the protective effect on the disruption of BSCB post-injury. In the current research, our results showed that treatment with NBP improved the locomotor function, prevented disruption of BSCB, and raised the expression of Occludin, β-Catenin, p120-Catenin and Claudin-5 which was thought to be the most vital membranous constituent of AJ and TJ. In vitro, TG, as a typical ER stress activator, increased paracellular permeability in HBMECs and disruption of AJ and TJ. NBP treatment reduced the degradation of AJ and TJ. Altogether, our findings suggested that NBP treatment shows the protective effect on the disruption of BSCB and improves locomotion recovery after injury. As far as I know, the present research, for the first time, suggested that NBP has protective effect on the integrity of BSCB following SCI.
There are numerous cellular and molecular mechanisms related to the BSCB permeability increasing induced by SCI. Reactive oxygen species (ROS) presents an ordinary initiate for numerous downstream paths that plays a critical role in BSCB dysfunction such as matrix metalloproteinases (MMP) activation, TJ modification and oxidative damage [37-39]. MMP-9 is involved in secondary damage induced by SCI and the disruption of BSCB by degrading the Occludin and Claudin-5 protein, rendering the leaky of BSCB and admitting neurotoxic components and immune cell to access the microenvironment of injured spinal cord. Inhibiting MMP activation by fluoxetine and valproic acidcan can inhibit the disruption of BSCB and improved locomotion recovery post-SCI [37, 40]. Inflammatory also play an important effect on the disruption of blood-CNS barrier under pathological disorder [41]. In our earlier research, we firstly validated that ER stress plays a vital role on the TJs regulation and BSCB disruption in the model of SCI [27, 42]. Inhibition of ER stress by PBA considerably prevented the increasing of BSCB permeability, and degradation of AJ and TJ proteins such as Occludin, β-Catenin, Claudin5 and p120-Catenin at 24 h post-SCI, thus, improved locomotion recovery. In addition, PBA prevents the degradation of AJ and TJ proteins via inhibiting ER stress in TG-treated cells [42]. In the current research, we verify a similar effect of NBP on trauma SCI and TG-treated HBMECs. We found that the permeability of BSCB was significantly increased at 24 h following SCI accompanying the stimulation of ER stress. In vitro, exogenously FITC-dextran past the monolayer was considerably increased compared to the control group by the treatment with TG. However, inhibition of ER stress by NBP significantly attenuated the permeability of BSCB via inhibiting the degradation of AJ and TJ protein in vitro and in vivo.
Earlier studies including our own demonstrate that ER stress shows a vital role in secondary damages following SCI [28] and inhibition of ER stress promotes functional recovery post-SCI [40, 43]. It is known that cells display a protective cellular signaling path via upregulating molecular chaperones expression at the period of early or middle ER stress [12]. However, when ER stress produced excessively, it may trigger an apoptosis path via initiation of CHOP and caspase-12 that induce cell apoptosis [44]. Growing research concentrate on ER stress in lots of cells (astrocytes, microglia, oligodendrocytes and neuron) after SCI has been done, but, the role of ER stress on the disruption of BSCB following SCI and the cellular mechanisms involved have not been investigated clearly. Although little evidence shows the role of ER stress on the disruption of BSCB after injury, recent research has presented that ER stress plays a significant part on the loss of AJ and TJ proteins in other barrier. In quiescent inflammatory bowel disease, ER stress induces the disruption of the intestinal epithelial barrier with an increase of permeability and a decrease of goblet cells number [45, 46]. Similarly, ER stress is involved in disrupting urothelial barrier-associated proteins (E-Cadherin, Claudin-4 and ZO-1) expression caused by ketamine [47]. GRP78 plays a vital role in human pulmonary artery endothelial cell injury and inflammation with the activation of NF-kB and vascular permeability regulation [48]. In addition, the retinal endothelial cell layer permeability is significantly increased through down-regulation of the expression of claudin-5 with TG treatment [49, 50]. All these results including our own both in vitro and in vivo experiments powerfully suggest ER stress shows the significant effect on TJ and AJ proteins expression. Whereas the precise function of ER stress on TJ proteins regulation keeps unclear in all kinds of ECs particularly about BSCB and further research is need.
NBP inhibits the degradation of AJ and TJ proteins and the expressions of ER stress-associated proteins in vitro. (A, B) Immunofluorescence staining of Claudin-5, p120-Catenin (green) in cells, nuclei is labeled with Hoechst (blue). TG treated with NBP inhibited the loss of p120-Catenin, Claudin-5. (C) Immunofluorescence staining of PDI (red) in endothelial cells, nuclei is marked by Hoechst (blue). NBP treatment reduced the expression of PDI compared with TG group. All experiments were performed in triplicate.
To confirm the signaling mechanisms of the dysfunction of ER following SCI and the effect of NBP on ER stress, we examined the levels of several ER stress-associated proteins and found that, as shown in Fig.3, the levels of EIF 2α/p- EIF 2α, ATF-4, ATF-6, CHOP, PDI, GRP78 and XBP-1 were boosted at 24h post-SCI. Moreover, NBP significantly attenuated the levels of ER stress associated proteins at 24h after SCI compared with SCI group. Similar findings were noted in cultures of EC cells subjected to TG stimulation (Fig. 6). Furthermore, NBP administration also reversed TG-induced increasing of the levels of cleaved caspase-12, leads to improvements in EC cell viability. The current findings display that ER stress shows the significant effect on BSCB disruption post-injury and that NBP inhibits BSCB disruption by regulating ER stress. Thus, adjustment of ER stress associated path may provide an underlying target for BSCB disruption in treatment of SCI.
NBP inhibits ER stress in TG-treated endothelial cells. Endothelial cells were treated with TG or TG + NBP for 6 h. (A, B) Typical western blots of ER stress related proteins ATF-6, ATF-4, PDI, XBP-1, GRP78, Cleaved-caspase12 and CHOP in each group cells. TG activated ER stress which was inhibited by NBP in endothelial cells. (C, D) Quantification data of ER stress associated proteins in each group cells. # P<0.05 versus the TG group, * P < 0.05 versus the Control group. All experiments were performed in triplicate.
NBP was synthesized and authorized for clinical application in patients of stroke in 2002 by China's State Food and Drug Administration [51]. Unlike preclinical drugs, its safety and stability have been repeatedly demonstrated in clinical practice. Furthermore, repurposing NBP may significantly reduce the costs and time associated with new drug development. The current research lays a foundation for future translational confidence of NBP in SCI, particularly the relations to the disruption of BSCB. Yet, much still remain to be done. Evaluate the NBP in different SCI animal models including rats and higher-level mammals including humans and old monkeys.
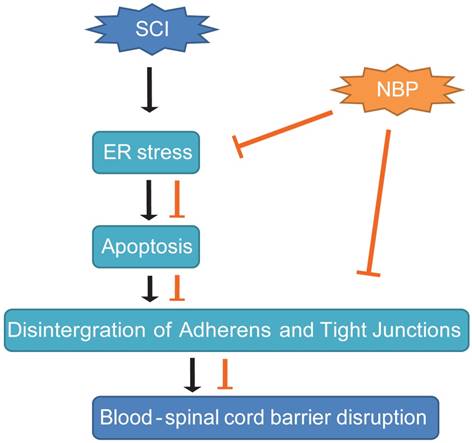
In conclusion, our study presented the proof that NBP obviously diminished BSCB permeability and breakdown of AJ and TJ proteins such as Occludin, p120-Catenin, β-Catenin and Claudin-5 at 24 h post-SCI, then, improved locomotion recovery. We first reported that the sheltering effect of NBP on BSCB was associated with the suppression of ER stress post-SCI. Furthermore, NBP considerably prevented the increase of the expression of ER stress-associated proteins and inhibited the depletion of TJ and AJ proteins in HBMECs treated with TG. These data indicate that therapeutic approach pointing on ER stress pathway may be applicable to the treatment of conserving the integrity of BSCB following SCI, and NBP, a novel agent for clinical treatment of stroke, appears to be promising drug for protecting the CNS neurological diseases characterized by a compromised BSCB (Fig. 8). However, in our research we did not clearly explain how NBP inhibits ER-stress induced apoptosis following SCI. In our further studies, we will concentrate on the specific mechanism of NBP on the inhibition of ER stress-induced apoptosis. Last but not the least, comprehending the active mechanism of NBP will contribute to the progress of more effective analogs for therapy of SCI.
A model exemplifying the protective effect of NBP on BSCB disruption following SCI. The activation of ER stress was involved in SCI, which led to damage the integrity of BSCB. ER stress-induced CHOP and caspase-12 activation resulted in the apoptosis of ECs after SCI, then accelerated the loss of AJ and TJ proteins. NBP treatment prevents the disruption of blood-spinal cord barrier via inhibiting endoplasmic reticulum stress following spinal cord injury.
Acknowledgements
This research was partly sustained by appropriation from National Natural Science Foundation of China (81572237, 81572227, 81501953), Technologies Science and technology Program of Zhejiang Province (2016C33107), Public Technology Research Program of Zhejiang Province (LGF18H060011, GF18H060032), Zhejiang Provincial Natural Science Foundation (Q16H090023, Y14H170002), Ningbo City Natural Science Foundation (2015A610213, 2015A610208), Science and Technology Planning Program of Taizhou City (1702KY04).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bartanusz V, Jezova D, Alajajian B, Digicaylioglu M. The blood-spinal cord barrier: morphology and clinical implications. Annals of neurology. 2011;70:194-206
2. Khayrullina G, Bermudez S, Byrnes KR. Inhibition of NOX2 reduces locomotor impairment, inflammation, and oxidative stress after spinal cord injury. Journal of neuroinflammation. 2015;12:172
3. Wu Q, Zhang YJ, Gao JY, Li XM, Kong H, Zhang YP. et al. Aquaporin-4 mitigates retrograde degeneration of rubrospinal neurons by facilitating edema clearance and glial scar formation after spinal cord injury in mice. Molecular neurobiology. 2014;49:1327-37
4. Karalija A, Novikova LN, Kingham PJ, Wiberg M, Novikov LN. The effects of N-acetyl-cysteine and acetyl-L-carnitine on neural survival, neuroinflammation and regeneration following spinal cord injury. Neuroscience. 2014;269:143-51
5. Santos-Nogueira E, Lopez-Serrano C, Hernandez J, Lago N, Astudillo AM, Balsinde J. et al. Activation of Lysophosphatidic Acid Receptor Type 1 Contributes to Pathophysiology of Spinal Cord Injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2015;35:10224-35
6. Sitia R, Braakman I. Quality control in the endoplasmic reticulum protein factory. Nature. 2003;426:891-4
7. Fu S, Watkins SM, Hotamisligil GS. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell metabolism. 2012;15:623-34
8. Zha BS, Zhou H. ER Stress and Lipid Metabolism in Adipocytes. Biochemistry research international. 2012;2012:312943
9. Liu XD, Ko S, Xu Y, Fattah EA, Xiang Q, Jagannath C. et al. Transient aggregation of ubiquitinated proteins is a cytosolic unfolded protein response to inflammation and endoplasmic reticulum stress. The Journal of biological chemistry. 2012;287:19687-98
10. Lin Z, Hu Y, Wang Z, Pan S, Zhang H, Ye L. et al. Intranasal basic fibroblast growth factor attenuates endoplasmic reticulum stress and brain injury in neonatal hypoxic-ischaemic injury. American journal of translational research. 2017;9:275-88
11. Zhang HY, Wang ZG, Lu XH, Kong XX, Wu FZ, Lin L. et al. Endoplasmic reticulum stress: relevance and therapeutics in central nervous system diseases. Molecular neurobiology. 2015;51:1343-52
12. Wang Z, Zhang H, Xu X, Shi H, Yu X, Wang X. et al. bFGF inhibits ER stress induced by ischemic oxidative injury via activation of the PI3K/Akt and ERK1/2 pathways. Toxicology letters. 2012;212:137-46
13. Briggs DI, Defensor E, Memar Ardestani P, Yi B, Halpain M, Seabrook G. et al. Role of Endoplasmic Reticulum Stress in Learning and Memory Impairment and Alzheimer's Disease-Like Neuropathology in the PS19 and APPSwe Mouse Models of Tauopathy and Amyloidosis. eNeuro. 2017:4
14. Zhang H, Wu F, Kong X, Yang J, Chen H, Deng L. et al. Nerve growth factor improves functional recovery by inhibiting endoplasmic reticulum stress-induced neuronal apoptosis in rats with spinal cord injury. Journal of translational medicine. 2014;12:130
15. Huang SQ, Tang CL, Sun SQ, Yang C, Xu J, Wang KJ. et al. Demyelination initiated by oligodendrocyte apoptosis through enhancing endoplasmic reticulum-mitochondria interactions and Id2 expression after compressed spinal cord injury in rats. CNS Neurosci Ther. 2014;20:20-31
16. Zhang Z, Tong N, Gong Y, Qiu Q, Yin L, Lv X. et al. Valproate protects the retina from endoplasmic reticulum stress-induced apoptosis after ischemia-reperfusion injury. Neuroscience letters. 2011;504:88-92
17. Li L, Zhang B, Tao Y, Wang Y, Wei H, Zhao J. et al. DL-3-n-butylphthalide protects endothelial cells against oxidative/nitrosative stress, mitochondrial damage and subsequent cell death after oxygen glucose deprivation in vitro. Brain research. 2009;1290:91-101
18. Chang Q, Wang XL. Effects of chiral 3-n-butylphthalide on apoptosis induced by transient focal cerebral ischemia in rats. Acta pharmacologica Sinica. 2003;24:796-804
19. Xu HL, Feng YP. Inhibitory effects of chiral 3-n-butylphthalide on inflammation following focal ischemic brain injury in rats. Acta pharmacologica Sinica. 2000;21:433-8
20. Huang JZ, Chen YZ, Su M, Zheng HF, Yang YP, Chen J. et al. dl-3-n-Butylphthalide prevents oxidative damage and reduces mitochondrial dysfunction in an MPP(+)-induced cellular model of Parkinson's disease. Neuroscience letters. 2010;475:89-94
21. Zhang T, Jia W, Sun X. 3-n-Butylphthalide (NBP) reduces apoptosis and enhances vascular endothelial growth factor (VEGF) up-regulation in diabetic rats. Neurological research. 2010;32:390-6
22. Lu YM, Huang JY, Wang H, Lou XF, Liao MH, Hong LJ. et al. Targeted therapy of brain ischaemia using Fas ligand antibody conjugated PEG-lipid nanoparticles. Biomaterials. 2014;35:530-7
23. Zhang Y, Wang L, Li J, Wang XL. 2-(1-Hydroxypentyl)-benzoate increases cerebral blood flow and reduces infarct volume in rats model of transient focal cerebral ischemia. The Journal of pharmacology and experimental therapeutics. 2006;317:973-9
24. Feng X, Peng Y, Liu M, Cui L. DL-3-n-butylphthalide extends survival by attenuating glial activation in a mouse model of amyotrophic lateral sclerosis. Neuropharmacology. 2012;62:1004-10
25. Zhang HY, Wang ZG, Wu FZ, Kong XX, Yang J, Lin BB. et al. Regulation of autophagy and ubiquitinated protein accumulation by bFGF promotes functional recovery and neural protection in a rat model of spinal cord injury. Molecular neurobiology. 2013;48:452-64
26. Wang HL, Lai TW. Optimization of Evans blue quantitation in limited rat tissue samples. Scientific reports. 2014;4:6588
27. Zhou Y, Zhang H, Zheng B, Ye L, Zhu S, Johnson NR. et al. Retinoic Acid Induced-Autophagic Flux Inhibits ER-Stress Dependent Apoptosis and Prevents Disruption of Blood-Spinal Cord Barrier after Spinal Cord Injury. International journal of biological sciences. 2016;12:87-99
28. Ohri SS, Maddie MA, Zhao Y, Qiu MS, Hetman M, Whittemore SR. Attenuating the endoplasmic reticulum stress response improves functional recovery after spinal cord injury. Glia. 2011;59:1489-502
29. Liu S, Sarkar C, Dinizo M, Faden AI, Koh EY, Lipinski MM. et al. Disrupted autophagy after spinal cord injury is associated with ER stress and neuronal cell death. Cell death & disease. 2015;6:e1582
30. Zheng B, Ye L, Zhou Y, Zhu S, Wang Q, Shi H. et al. Epidermal growth factor attenuates blood-spinal cord barrier disruption via PI3K/Akt/Rac1 pathway after acute spinal cord injury. Journal of cellular and molecular medicine. 2016;20:1062-75
31. Matsushita T, Lankford KL, Arroyo EJ, Sasaki M, Neyazi M, Radtke C. et al. Diffuse and persistent blood-spinal cord barrier disruption after contusive spinal cord injury rapidly recovers following intravenous infusion of bone marrow mesenchymal stem cells. Experimental neurology. 2015;267:152-64
32. Lee JY, Choi HY, Ahn HJ, Ju BG, Yune TY. Matrix metalloproteinase-3 promotes early blood-spinal cord barrier disruption and hemorrhage and impairs long-term neurological recovery after spinal cord injury. The American journal of pathology. 2014;184:2985-3000
33. Winkler EA, Sengillo JD, Sagare AP, Zhao Z, Ma Q, Zuniga E. et al. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E1035-42
34. Wu J, Ling J, Wang X, Li T, Liu J, Lai Y. et al. Discovery of a potential anti-ischemic stroke agent: 3-pentylbenzo[c]thiophen-1(3H)-one. Journal of medicinal chemistry. 2012;55:7173-81
35. Li J, Li Y, Ogle M, Zhou X, Song M, Yu SP. et al. DL-3-n-butylphthalide prevents neuronal cell death after focal cerebral ischemia in mice via the JNK pathway. Brain research. 2010;1359:216-26
36. Hu J, Wen Q, Wu Y, Li B, Gao P. The effect of butylphthalide on the brain edema, blood-brain barrier of rats after focal cerebral infarction and the expression of Rho A. Cell biochemistry and biophysics. 2014;69:363-8
37. Lee JY, Kim HS, Choi HY, Oh TH, Yune TY. Fluoxetine inhibits matrix metalloprotease activation and prevents disruption of blood-spinal cord barrier after spinal cord injury. Brain: a journal of neurology. 2012;135:2375-89
38. Yun HM, Park KR, Kim EC, Hong JT. PRDX6 controls multiple sclerosis by suppressing inflammation and blood brain barrier disruption. Oncotarget. 2015;6:20875-84
39. Kiyatkin EA, Sharma HS. Breakdown of Blood-Brain and Blood-Spinal Cord Barriers During Acute Methamphetamine Intoxication: Role of Brain Temperature. CNS & neurological disorders drug targets. 2016;15:1129-38
40. Lee JY, Maeng S, Kang SR, Choi HY, Oh TH, Ju BG. et al. Valproic Acid Protects Motor Neuron Death by Inhibiting Oxidative Stress and Endoplasmic Reticulum Stress-Mediated Cytochrome C Release after Spinal Cord Injury. Journal of neurotrauma. 2014
41. Yu DS, Cao Y, Mei XF, Wang YF, Fan ZK, Wang YS. et al. Curcumin improves the integrity of blood-spinal cord barrier after compressive spinal cord injury in rats. Journal of the neurological sciences. 2014;346:51-9
42. Zhou Y, Ye L, Zheng B, Zhu S, Shi H, Zhang H. et al. Phenylbutyrate prevents disruption of blood-spinal cord barrier by inhibiting endoplasmic reticulum stress after spinal cord injury. American journal of translational research. 2016;8:1864-75
43. Zhu SP, Wang ZG, Zhao YZ, Wu J, Shi HX, Ye LB. et al. Gelatin Nanostructured Lipid Carriers Incorporating Nerve Growth Factor Inhibit Endoplasmic Reticulum Stress-Induced Apoptosis and Improve Recovery in Spinal Cord Injury. Molecular neurobiology. 2015
44. Zhang HY, Zhang X, Wang ZG, Shi HX, Wu FZ, Lin BB. et al. Exogenous basic fibroblast growth factor inhibits ER stress-induced apoptosis and improves recovery from spinal cord injury. CNS Neurosci Ther. 2013;19:20-9
45. McGuckin MA, Eri RD, Das I, Lourie R, Florin TH. Intestinal secretory cell ER stress and inflammation. Biochemical Society transactions. 2011;39:1081-5
46. Vivinus-Nebot M, Frin-Mathy G, Bzioueche H, Dainese R, Bernard G, Anty R. et al. Functional bowel symptoms in quiescent inflammatory bowel diseases: role of epithelial barrier disruption and low-grade inflammation. Gut. 2014;63:744-52
47. Liu KM, Chuang SM, Long CY, Lee YL, Wang CC, Lu MC. et al. Ketamine-induced ulcerative cystitis and bladder apoptosis involve oxidative stress mediated by mitochondria and the endoplasmic reticulum. American journal of physiology Renal physiology. 2015;309:F318-31
48. Leonard A, Paton AW, El-Quadi M, Paton JC, Fazal F. Preconditioning with endoplasmic reticulum stress ameliorates endothelial cell inflammation. PloS one. 2014;9:e110949
49. Adachi T, Yasuda H, Nakamura S, Kamiya T, Hara H, Hara H. et al. Endoplasmic reticulum stress induces retinal endothelial permeability of extracellular-superoxide dismutase. Free radical research. 2011;45:1083-92
50. Adachi T, Teramachi M, Yasuda H, Kamiya T, Hara H. Contribution of p38 MAPK, NF-kappaB and glucocorticoid signaling pathways to ER stress-induced increase in retinal endothelial permeability. Arch Biochem Biophys. 2012;520:30-5
51. Wang YG, Li Y, Wang CY, Ai JW, Dong XY, Huang HY. et al. L-3-n-Butylphthalide protects rats' cardiomyocytes from ischaemia/reperfusion-induced apoptosis by affecting the mitochondrial apoptosis pathway. Acta physiologica (Oxford, England). 2014;210:524-33
Author contact
![]() Corresponding authors: Jian Xiao, School of Pharmacy, Wenzhou Medical University, Wenzhou 325035, Zhejiang 325035, PR China. Huazi Xu, Department of Orthopaedics, The Second Affiliated Hospital, Wenzhou Medical University, Wenzhou 325000, PR China. Tel.: +86-577-88816381 Fax.: +86-577-85773087 E-mail: xfxj2000com; spine-xucom.
Corresponding authors: Jian Xiao, School of Pharmacy, Wenzhou Medical University, Wenzhou 325035, Zhejiang 325035, PR China. Huazi Xu, Department of Orthopaedics, The Second Affiliated Hospital, Wenzhou Medical University, Wenzhou 325000, PR China. Tel.: +86-577-88816381 Fax.: +86-577-85773087 E-mail: xfxj2000com; spine-xucom.