Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Methods and Materials
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(5):1042-1051. doi:10.7150/ijbs.31099 This issue Cite
Research Paper
miR-30e-5p Mitigates Hypoxia-Induced Apoptosis in Human Stem Cell-Derived Cardiomyocytes by Suppressing Bim
Binhai Mo, Xiaodan Wu, Xiantao Wang, Jian Xie, Ziliang Ye, Lang Li ![]()
Department of cardiology, The First Affiliated Hospital of Guangxi Medical University & Guangxi Key Laboratory Base of Precision Medicine in Cardio-cerebrovascular Diseases Control and Prevention & Guangxi Clinical Research Center for Cardio-cerebrovascular Diseases, Nanning 530021, China
Received 2018-10-31; Accepted 2019-3-6; Published 2019-4-21
Abstract

Coronary microembolization can cause slow or no reflow, which is one of the crucial reasons for reverse of clinical advantage from cardiac reperfusion therapy. miRNAs and apoptosis are dramatically involved in the occurrence and process of cardiovascular diseases. Fortunately, human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) have emerged as an appealing model for the evaluation of cardiovascular diseases. Therefore, our study was designed to explore the role of miR-30e-5p and apoptosis in a hypoxia-induced hiPSC-CM injury model. Our results showed that the expression levels of miR-30e-5p were overtly downregulated in a time-dependent manner under hypoxic conditions. Expression of miR-30e-5p was significantly downregulated after 24 hours of hypoxia, hypoxia treatment dramatically induced apoptosis. Calcium handling capability significantly decreased after 24 hours of hypoxia treatment. miR-30e-5p overexpression partially mitigated hypoxia-induced apoptosis and rescued hypoxia-induced calcium handling defects in hiPSC-CMs. The luciferase reporter assay showed that miR-30e-5p can directly target the 3'-UTR of Bim, which is an apoptosis activator and autophagy suppressor. The mRNA and protein of Bim remarkably increased after hypoxia treatment and reduced with miR-30e-5p overexpression. Moreover, downregulation of Bim mitigated hypoxia-induced apoptosis and activated autophagy. These results demonstrated that miR-30e-5p mitigated hypoxia-induced apoptosis in hiPSC-CMs at least in part via Bim suppression and subsequent autophagy activation. Our study suggested miR-30e-5p may act as a potential therapeutic target for coronary microembolization.
Keywords: microRNA-30e, iPSCs, cardiomyocytes, apoptosis, autophagy, CME
Introduction
Coronary microembolization (CME) is considered a leading complication during percutaneous coronary intervention (PCI)[1]. CME can result in slow or no reflow. Optimal cardiac reperfusion cannot be repaired in nearly 65% of patients with prompt and efficient recanalization of epicardial culprit vessels. CME may also lead to cardiac contractile disfunction and myocardial arrhythmias, which are heavily related to the progression of infarct size and worsened patient condition at follow-up[2, 3]. Recently, microRNAs (miRNAs) have been implicated in the development of myocardial autophagy during CME [4, 5]; however, the underlying mechanism of these factors has not yet been fully elucidated.
miRNAs are endogenous noncoding RNAs of 18-24 nucleotides that can negatively regulate gene expression at the posttranscriptional level via pair- bonding with the 3'-UTR of a target mRNA[6]. Since one miRNA can target multiple genes, it has been found that miRNAs can participate in the regulation of diverse biochemical and physiological activities[6]. Increasing evidence shows that miRNAs have diagnostic and therapeutic potential in cardiovascular diseases [7]. Hypoxia can critically modify the miRNA profile in cardiomyocytes [8]. Previous studies have also reported that autophagy is related to the occurrence and process of most cardiovascular diseases [9]. Nevertheless, little evidence currently available has shown activated autophagy in cardiomyocytes during early myocardial hypoxia with treatment [10]. In a previous study, we found several miRNAs that were markedly downregulated in rat cardiomyocytes after microsphere infusion. One of those miRNAs was miR-30e-5p, a miRNA that may participate in cardiomyocyte apoptosis and autophagy. The miR-30 family has recently been reported to participate in autophagy regulation [11, 12]. However, the role of miR-30e-5p in the development of cardiomyocyte apoptosis and autophagy after CME remains unclear.
Cardiomyocytes are terminally differentiated cells and study of cardiomyocytes is hampered by the complexities in obtaining human heart tissue and the difficulties to propagate cardiac samples in culture. Consequently, most studies reported have used animal models to mimic cardiovascular disease. However, crucial biological and pathological differences exist between animal and human cardiac tissue, especially in cardiomyocytes. Recently, human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) have emerged as an appealing model for investigating cardiovascular diseases[13]. Generation of hiPSC-CMs offers an innovative platform for direct assessment of cardiomyocyte function in vitro[14]. Therefore, we utilized a novel hiPSC-CM model to mimic the development of hypoxia-induced injury during CME. Subsequently, the effect of miR-30e-5p on hiPSC-CM apoptosis and autophagy was investigated.
Methods and Materials
Generation of hiPSC-CMs
Human induced pluripotent stem cells (hiPSCs (derived from exfoliated renal epithelial cells; Cellapy, Beijing, China)) were employed in this study. The hiPSCs were cultured in human PSCeasy Medium (Cellapy, Beijing, China) on Matrigel (Corning, Bedford, MA). At 80% confluence, the cells were regularly passaged every 4 days using EDTA. The hiPSCs were cultured at 37°C with 5% CO2.
The hiPSC-CMs were derived from hiPSCs utilizing a small-molecule-based monolayer method as previously reported[15, 16]. Briefly, for the first 48 hours (day 0 to day 2), the hiPSCs were treated with 6 µM CHIR99021 (GSK-3 inhibitor; Selleck Chemical, USA) in basal medium (RPMI plus B27 without insulin (both from Invitrogen, USA)). For the second 48 hours (day 2 to day 4), the medium was replaced with basal medium. For the third 48 hours (day 4 to day 6), the medium was replaced with fresh basal medium with the inhibitor Wnt protein 2 (Selleck Chemical, USA). From day 7, the cells were maintained in the maintenance medium (RPMI plus regular B27; Invitrogen, USA), with the medium regularly replaced every other day to limit pH changes. Compared with fetal bovine serum (FBS), the RPMI plus B27 maintenance medium has the capacity to limit fibroblast overgrowth. The hiPSC-CMs 30 days after differentiation were employed in this study.
Cell transfection
Concentrated miR-30e-5p expression and negative control recombinant lentiviruses were obtained from Hanbio (Shanghai, China). hiPSC-CMs were separately treated with miR-30e-5p mimic or negative control lentivirus at MOI 3. Transduction medium was replaced with fresh RPMI supplemented with regular B27 after 24 h of incubation, and the cells were transfected with miRs. Three days after transfection, miR-30e-5p expression was assessed via RT-qPCR, and the cells were subsequently subjected to hypoxia treatment. Bim small interfering RNA (shBim) and negative control siRNA (shNC) recombinant lentiviruses were also obtained from Hanbio (Shanghai, China). hiPSC-CMs were separately treated with lentivirus Bim siRNA at 3 MOI or equivalent negative control siRNA empty vectors. Transfection medium was changed after 24 h of incubation and the subsequent hypoxia treatment.
Stimulated hypoxia-induced injury
hiPSC-CMs were exposed to four conditions: a normoxic group (N group), hypoxia group (H group), negative control group (NC+H group), and miRNA-30e-5p overexpression group (miR+H group). The hiPSC-CMs were incubated in starvation medium (glucose-free RPMI+ supplemented with regular B27) and then placed in a modular incubator chamber (Billups Rothenberg, Del Mar, CA, USA) flushed with a gas mixture to induce hypoxia conditions (1% O2/5% CO2/94% N2) at 37˚C for 24 h. The cells under normoxic culture conditions (5% CO2/95% air) served as the controls.
Caspase-3 activity assay
Caspase-3 activity was analyzed using a Caspase-3 assay kit (BioVision Inc., Mountain View, CA), according to the manufacturer's instructions. Briefly, hiPSC-CMs were first seeded in 6-well plates. After the cells were subjected to hypoxia conditions, Caspase-3 activity of the cells was analyzed according to the manufacturer's instructions. Finally, Caspase-3 activity was spectrophotometrically analyzed via absorbance measurements at 405 nm applying a Multi-Mode Reader (BioTek, Winooski, VT, USA).
Immunostaining and imaging analyses
Briefly, the cell samples were first fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 and blocked with 3% bovine serum albumin (BSA). After 3 rinses with PBS for 5 min, the samples were incubated with primary antibodies against cTnT (1:100; Santa Cruz Biotechnology, USA) and α-actinin (1:100; Santa Cruz Biotechnology, USA) overnight at 4˚C. After being rinsed 3 times with PBS, secondary goat anti-mouse IgG Alexa Fluor594 and goat anti-rabbit IgG Alexa Fluor488 (1:1000; Invitrogen, USA) antibodies were applied for 1 hour at room temperature in the dark. Nuclei of all samples were stained with 4,6-diamidino-2-phenylindole (DAPI; Abcam, USA) for 5 min. Fluorescence images were captured with Leica TCS SP5 MP confocal laser scanning microscope (Leica, Wetzlar, Germany).
RNA extraction and RT-qPCR
Total RNA samples were extracted using TRIzol reagent (TaKaRa, Japan) according to the manufacturer's instructions. The concentration of RNA was detected utilizing a NanoDrop spectrophotometer (Thermo Fisher Scientific Inc., USA), and then, the RNA was reverse-transcribed to cDNA using a cDNA reverse transcription kit (TaKaRa, Japan) according to the protocol supplied by the manufacturers. The gene expression levels were determined by RT-qPCR using a SYBR Green I PCR kit (TaKaRa, Japan) on an iCycler iQ5 system (Bio-Rad, USA). U6 DNA served as the miRNA internal control. The relative quantification was analyzed according to the 2-∆∆Ct method. Sequences of the primers were designed as follows: miR-30e-5p (forward: 5'- TGTAAACATCCTTGACTGGAAGG-3' and reverse: 5'-CCAGTGCGAATACCTCGGAC-3'); U6 (forward: 5'-GGTCGGGCAGGAAAGAGGGC-3' and reverse: 5'-GCTAATCTTCTCTGTATCGTTCC-3'); Bim (forward: 5'-CAAGAGTTGCGGCGTATTGGAG-3' and reverse: 5'-ACACCAGGCGGACAATGTAACG-3'). GAPDH (forward: 5'- GTCTCCTCTGACTTCAACAGCG-3' and reverse: 5'- ACCACCCTGTTGCTGTAGCCAA-3').
Flow cytometry analysis
The rate of apoptosis was examined via flow cytometry analysis (BD, USA) using an AnnexinV/PI Apoptosis Detection Kit (BD, USA) according to the manufacturer's instructions. hiPSC-CMs were first collected on day 30 of differentiation, dissociated employing CardioEasy cardiomyocyte dissociation enzyme (Cellapy, China), filtered softly through a 40-µm cell strainer and fixed with fixation buffer (BD, USA) for 15 min. Fixed cardiomyocytes were next washed with Wash buffer (BD, USA) and incubated with primary antibodies against cTnT (1:100; Santa Cruz Biotechnology) for 30 min at room temperature. The cardiomyocytes were then incubated with a Alexa Fluor 488-conjugated anti-mouse secondary antibody (1:100; Invitrogen) for 30 min. The cardiomyocytes were finally washed and measured via FACS analysis. The analysis was completed by the FlowJo program.
Western blotting
hiPSC-CMs were lysed using a Protein Extraction Kit (Solarbio, China). Protein concentrations of each sample was detected with a bicinchoninic acid (BCA) protein assay (Solarbio, China). Equal amounts (50 μg) of total protein samples were subjected to 10% SDS-PAGE and electrophoretically transferred onto a nitrocellulose membrane (Merck Millipore, USA). Then, the membranes were blocked with 5% nonfat milk in TBS for 1 h at room temperature and incubated with the indicated primary antibodies (anti-Bcl-2, anti-Bax, anti-Caspase-3, anti-Bim, anti-Beclin-1, anti-p62 and anti-GAPDH antibodies (all from Abcam, USA)) overnight at 4°C, respectively. GAPDH was served as an internal control. The membranes were then washed with TBST 3 times and incubated with the indicated infrared secondary antibodies (Invitrogen, USA) in the dark for 2 h at room temperature. The protein band intensities were finally quantified with an Odyssey infrared fluorescence scanning imaging system (Odyssey, LICOR, USA).
Luciferase reporter assay
The wild-type Bim 3'-UTR was amplified from human genomic DNA by PCR and cloned into a psiCHECK2 vector (Promega, USA). Mutation of the putative miR-30e-5p target site in the Bim 3'-UTR was obtained applying a Quick Change Site-Directed Mutagenesis Kit (Stratagene, Germany). For the luciferase reporter assay, HEK293T cells plated in 96-well plates were cotransfected with 160 ng of the Bim 3'-UTR reporter plasmids, 20 ng of Renilla luciferase report vector (Promega, USA), and 5 pmol miR-30e-5p mimic or negative control miRNA. At 48 h after transfection, the cells harvested were lysed and luciferase activities were detected applying a Dual-luciferase Reporter Assay System (Promega, USA). Firefly luciferase activities were finally normalized to corresponding Renilla luciferase.
Calcium imaging
hiPSC-CMs were dissociated, seeded in Matrigel-coated 8-well LAB-TEK II chambers (Nalgene Nunc International, USA) and allowed to recover from passage for three days. The hiPSC-CMs were then loaded with 10 mM Fluo-4 AM in Tyrode's solution and calcium signaling was acquired via confocal microscope (Carl Zeiss, LSM 700, Germany). Spontaneous calcium transients were imaged utilizing standard line-scan methods at a speed of 2.55 ls/pixel, with 8000 line scans.
Statistical analysis
The data are presented as the mean ± standard deviation. Statistical analysis was performed utilizing SPSS 20.0 software (SPSS, Inc., USA). Student's t-test was used to evaluate the significance of differences between two groups. Values of P<0.05 were considered to indicate statistical significance.
Results
Generation of hiPSC-CMs
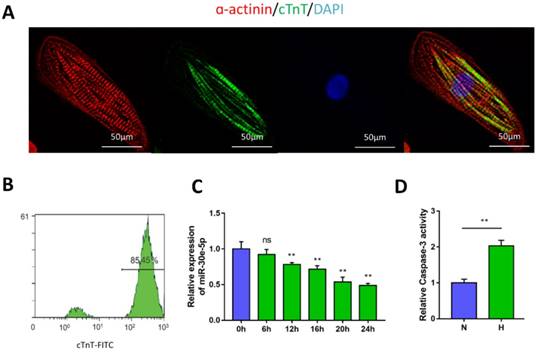
To generate cardiomyocytes from human pluripotent stem cells, a small-molecule-based monolayer differentiation method was used to induce differentiation. Some cells contract spontaneously from 7 days after induction, almost 90% of the cells beat vigorously after 10 days, with a beating rate of up to 100 beats per min (Movie S1 and Movie S2). To validate the efficiency of cardiomyocyte differentiation, immunostaining and flow cytometry were performed to examine the cardiomyocyte marker cTnT and α-actinin in hiPSC-CMs (Fig. 1A). The results showed the presence of cTnT in over 85% of the total cells (Fig. 1B).
Characterization of hiPSC-CMs and hypoxia-induced downregulation of miR-30e-5p. (A) Immunostaining of hiPSC-CMs after 30 days of differentiation and culture: α-actinin (red), cTnT (green), DAPI (blue). (B) Representative flow cytometry result showing the efficiency of hiPSC-CM differentiation. (C) Dynamic changes in miR-30e-5p expression determined by RT-qPCR in hiPSC-CMs treated with hypoxia. (D) Measurement of hiPSC-CM viability using Caspase-3 activity assay after the indicated treatments. All the data are expressed as the means ± S.E.M. Statistically significant differences between individual groups (n≥3; *P<0.05, **P<0.01, and ***P<0.001. Scale bar: 50µm).
Hypoxia-induced downregulation of miR-30e-5p triggered apoptosis in hiPSC-CMs
To determine the potential involvement of miR-30e-5p in CME, we first utilized an ex-vivo hypoxia model of CME in hiPSC-CMs and validated miR-30e-5p levels using RT-qPCR. Compared with the 0 h group (P<0.01), the expression levels of miR-30e-5p were overtly downregulated in a time-dependent manner, reaching a maximal suppression of 50% after 24 h of exposure to hypoxic conditions (Fig. 1C). Next, we detected whether miR-30e-5p downregulation is related to the hypoxia-induced apoptotic response in hiPSC-CMs. Flow cytometry analysis suggested that the ratio of apoptotic cells significantly increased in response to 24 h of hypoxia exposure along with Caspase-3 activity (Fig. 2B, 2C and 2D). Taken together, these data suggest that hypoxia may inhibit miR-30e-5p expression and boost hiPSC-CM apoptosis.
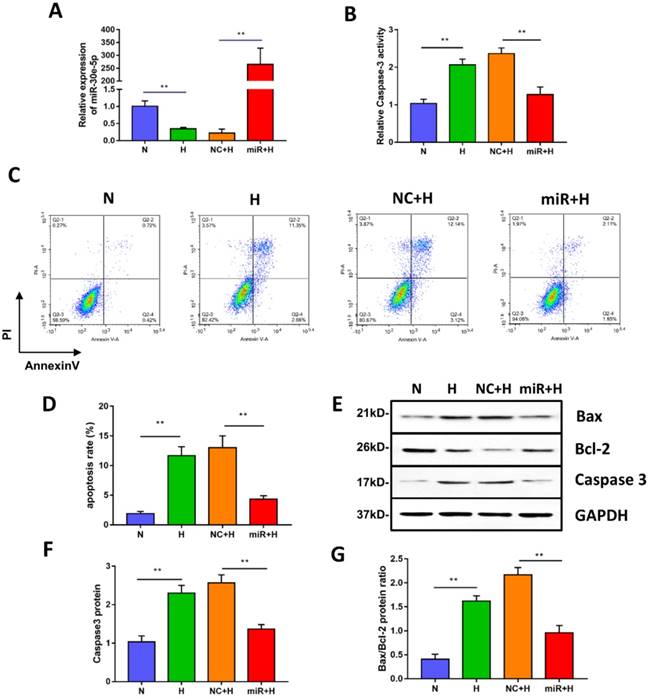
miR-30e-5p overexpression inhibited hypoxia-induced apoptosis in hiPSC-CMs. (A) RT-qPCR analysis of miR-30e-5p levels in hiPSC-CMs with the indicated treatments. (B) Measurement of hiPSC-CM viability using Caspase-3 activity assay after the indicated treatments. (C) Representative flow cytometry analysis of hiPSC-CMs after annexin-V/PI staining. (D) Quantification of cells positive for annexin-V/PI. (E) Western blot analysis of Bax,Bcl-2, and Caspase-3 protein levels. (F) Quantification of the Caspase-3 protein level. (G) Quantification of the Bax/Bcl-2 protein ratio. (n≥3; *P<0.05, **P<0.01, and ***P<0.001).
miR-30e-5p overexpression mitigated hypoxia-induced hiPSC-CM apoptosis
To investigate whether miR-30e-5p overexpression could mitigate apoptosis induced by hypoxia, we transiently transfected hiPSC-CMs with miR-30e-5p mimic or the equivalent negative control, and then, the transfection efficiency was validated by RT-qPCR. As shown in Fig. 2A, miR-30e-5p mimic successfully enhanced the expression level of miR-30e-5p in hiPSC-CMs at 72 h after transfection. We then performed Caspase-3 activity assay to assess cell viability and found that miR-30e-5p overexpression ameliorated the hypoxia-induced cell viability decrease at 24 h. Transfection with miR-negative control showed no effect on cell viability compared with the H group under hypoxia conditions (Fig. 2B). Consistent with the Caspase-3 activity assay results, overexpression of miR-30e-5p significantly attenuated apoptosis in hiPSC-CMs subjected to 24 h of hypoxia (Fig. 2C and 2D). In contrast, transfection with the miR-negative control (NC+H) had no significant effect on hypoxia- induced apoptosis. The number of apoptotic cells stained positive for annexin-V/PI was examined via flow cytometry and was found to be reduced from 15% (NC+H) to 5% (miR+H) (P<0.01). Furthermore, Caspase-3 levels were decreased in the miR-30e-5p overexpression group (P<0.01) (Fig. 2E and 2F).
To determine the effect of miR-30e-5p overexpression on the ratio of Bax/Bcl-2 in hiPSC-CMs exposed to hypoxia, we performed a Western blotting analysis, which showed that hiPSC-CMs transfected with miR-30e-5p mimic presented a lower Bax/Bcl-2 ratio under both normoxic and hypoxic conditions, accompanied by a decrease in the Caspase-3 level (Fig. 2E, 2F and 2G). These data indicate that miR-30e-5p might have cytoprotective effects via suppression of hiPSC-CM apoptosis in response to hypoxia.
miR-30e-5p overexpression rescued hypoxia-induced calcium handling defects in hiPSC-CMs
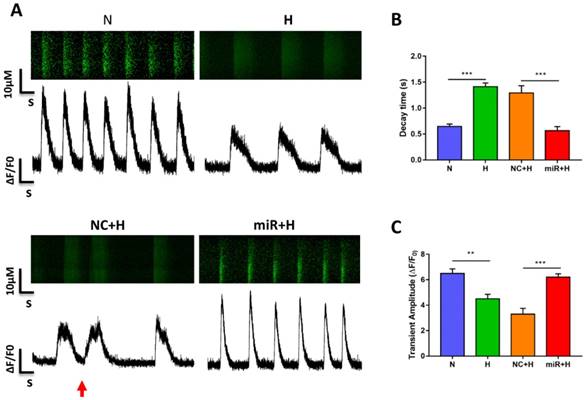
To examine the physiological impact of miR-30e-5p on hiPSC-CMs after hypoxia-induced apoptosis, we analyzed the Ca2+ handling properties of hiPSC-CMs after hypoxia injury using the fluorescent Ca2+ dye Fluo-4 acetoxymethyl ester (AM). Hypoxia impaired excitation-contraction coupling in the hiPSC-CMs was demonstrated as a decline in the amplitude of transients and decay time as well as the occurrence of irregular transients. However, overexpression of miR-30e-5p significantly enhanced transient amplitude and shortened the decay time (Fig. 3A-C). Abnormal Ca2+ transients, such as block-like transients were also present in the H group and NC+H group but not in the N and miR+H groups (Fig. 3A). These findings demonstrate that miR-30e-5p might rescue Ca2+ handling properties in hiPSC-CMs after hypoxia-induced injury.
Assessment of Ca2+ handling properties in hiPSC-CMs. (A) Representative Ca2+ line scans and waveforms in hiPSC-CMs. Red arrowheads indicate abnormal Ca2+ waveforms. (B) Quantification of decay time in hiPSC-CMs. (C) Mean transient amplitudes of ∆F/F0 ratios in hiPSC-CMs (n=30; *P<0.05, **P<0.01, and ***P<0.001).
miR-30e-5p overexpression inhibited the expression of the apoptosis activator Bim
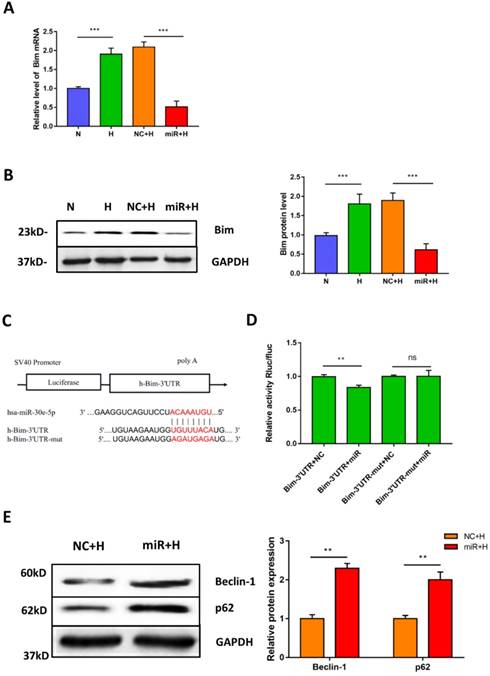
To elucidate the potential mechanism of miR-30e-5p in the regulation of hypoxia-induced apoptosis. We first identified the putative targets of miR-30e-5p, via in silico analysis using miRanda (http://www.microrna.org) and TargetScan (http://www.targetscan.org). We found that the 3'-UTR of Bim contains one potential target site for miR-30e-5p that is highly conserved among vertebrates. Therefore, we hypothesized that miR-30e-5p might attenuate hypoxia-induced apoptosis in hiPSC-CMs by suppressing Bim expression. To examine this hypothesis, we assessed the mRNA levels of Bim after hypoxia treatment. As expected, hypoxia remarkably upregulated Bim in hiPSC-CMs compared with cells in the N group. Moreover, transfection with miR-30e-5p significantly suppressed Bim expression in hiPSC-CMs after 24 h of hypoxia treatment compared with the NC+H group (Fig. 4A). In addition, hiPSC-CMs overexpressing miR-30e-5p did not exhibit Bim protein upregulation after hypoxia treatment (Fig. 4B). To validate whether miR-30e-5p directly targets the 3'-UTR of Bim, we inserted the 3'-UTR of Bim or a mutated sequence into a luciferase reporter and transiently cotransfected HEK-293 cells with either miR-30e-5p mimic or miR-negative(Fig. 4C). Luciferase reporter assay data showed that miR-30e-5p inhibited Bim translation via direct binding to the 3'-UTR of Bim (P<0.01) (Fig. 4D). Taken together, these results indicate that miR-30e-5p can mediate Bim expression during hypoxia at the posttranscriptional level.
Bim is a direct target of miR-30e-5p in hiPSC-CMs. (A) RT-qPCR analysis of Bim levels in hiPSC-CMs. (B) Western blot analysis of Bim protein. (C)Schematic representation of miR-30e-5p target sequences and Bim 3'-UTR luciferase reporter constructs. (D) The relative luciferase activity in luciferase reporter assays. (E) Level of the autophagy-related proteins Beclin-1 and p62 in hiPSC-CMs after 24 h of hypoxia treatment. (n≥3; *P<0.05, **P<0.01, and ***P<0.001).
Downregulation of Bim mitigated hypoxia-induced apoptosis and activated autophagy in hiPSC-CMs
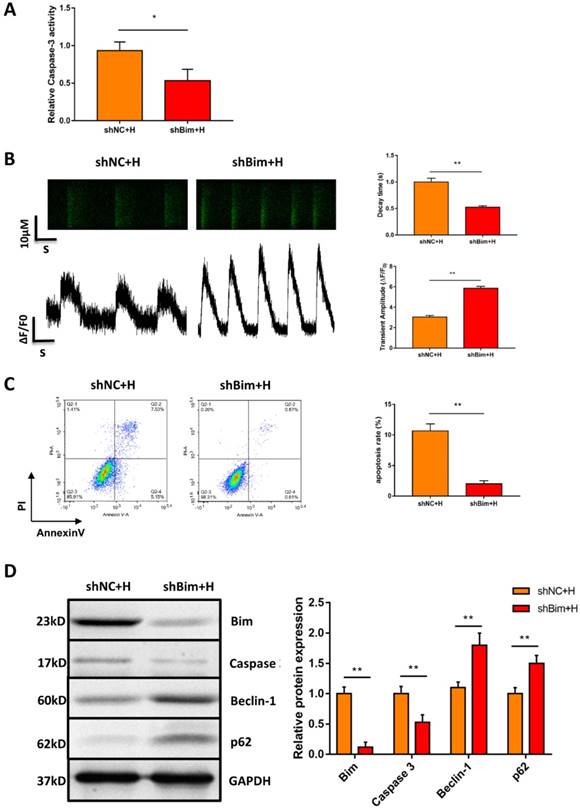
Considering that Bim also acts as an autophagy suppressor in addition to activating apoptosis, we explored whether downregulation of Bim promoted autophagy activation mediated by miR-30e-5p overexpression. The levels of the autophagy-related proteins Beclin-1 and p62 were measured via Western blotting. Our data showed that downregulation of Bim enhanced the levels of Beclin-1 and p62 in hiPSC-CMs overexpressing miR-30e-5p compared with the NC+H group (Fig. 4E). To further confirm that miR-30e-5p protected hiPSC-CMs against hypoxia-induced apoptosis by inhibiting Bim expression, we knocked down the expression of Bim via siRNA technology. Downregulation of Bim resulted in reduction of Caspase-3 activity (Fig. 5A) and elevation of Ca2+ handling properties (Fig. 5B). Additionally, The number of apoptotic cells stained positive for annexin-V/PI was examined via flow cytometry and was found to be reduced from 12% (shNC+H) to 2% (shBim+H) (Fig. 5C). Western blotting analysis showed that Caspase-3 proteins expression reduced and autophagy-related proteins expression increased in hiPSC-CMs (Fig. 5D). Hence, these results suggest that Bim downregulated might mitigate hypoxia-induced apoptosis and activate autophagy in hiPSC-CMs.
Downregulation of Bim mitigated hypoxia-induced apoptosis and activated autophagy in hiPSC-CMs. (A) Measurement of hiPSC-CM viability using Caspase-3 activity assay after the indicated treatments. (B) Assessment of Ca2+ handling properties in hiPSC-CMs. (C) Representative flow cytometry analysis of hiPSC-CMs after Bim knockdown. (D) Level of Bim protein, Caspase-3 protein and autophagy-related proteins in hiPSC-CMs after Bim knockdown. H: hypoxia; shNC: Bim knockdown-negative control; shBim: Bim knockdown. (n=3; **P<0.01).
Discussion
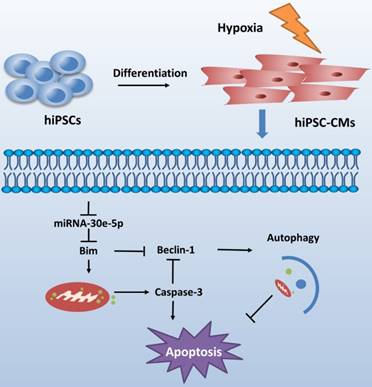
CME is now defined as a serious complication that diminishes the potential value of reperfusion therapy. Coronary microinfarction and microcirculation thrombosis are two primary pathogenic components involved in the occurrence of CME when atherosclerotic plaques rupture during PCI[17]. Coronary distal microembolization may cause relentless hypoxia in cardiovascular microcirculation and consequently myocardial injury followed by poor ventricular remodeling. Despite efforts to elucidate the mechanisms of ischemia/reperfusion injury in the CME process [18-20], the mechanisms underlying the development of distal microembolization remain unclear. hiPSC-CMs have been developed as an appealing model for the evaluation of cardiovascular diseases[13]. In this study, we developed a novel hiPSC-CM model to mimic the development of hypoxia-induced injury during CME. We initially identified a significant downregulation of miR-30e-5p in hiPSC-CMs exposed to hypoxia and further determined that miR-30e-5 overexpression protected hiPSC-CMs against hypoxia-induced apoptosis via direct targeting of Bim. Remarkably, downregulation of Bim prevented apoptosis and activated autophagy in hiPSC-CMs after hypoxia treatment (Fig. 6).
Schematic shows proposed mechanism for miR-30e-5p mediating hypoxia-induced apoptosis in hiPSC-CMs. miR-30e-5p, downregulated by hypoxia, inhibits hypoxia-induced hiPSC-CMs apoptosis by suppression of Bim protein, an apoptosis activator and autophagy repressor, thereby activating autophagy and apoptosis inhibition.
miR-30e-5p is a member of the miR-30 family, which contains five individual miRNAs, namely, miR-30a, miR-30b, miR-30c, miR-30d and miR-30e. The miR-30 family are abundantly expressed in heart tissues, which extensively contribute to the pathogenesis of multiple cardiovascular diseases [21-23]. Recently, studies on the miR-30 family are mostly related to heart failure, cardiac infarction, myocardial hypertrophy and ischemia/ reperfusion injury [24, 25]. In addition, one study showed that the miR-30 family also performs a vital role in attenuation of myocardial ischemic injury[26]. Our work revealed that miR-30e-5p expression is markedly downregulated 24 h after hypoxia-induced apoptosis, while autophagy is significantly suppressed.
Bim, an apoptosis activator belonging to the Bcl-2 protein family, is a potential target of miR-30e-5p. Dysregulation of Bim is involved in oncogenesis and chemo-resistance of solid tumors[27, 28]. As a pro-apoptotic regulator, Bim induces apoptosis through negating the Bcl-2 protein family and direct activating Bax/Bak oligomerization[29]. In primed pluripotent stem cells, a previous study demonstrated that inhibition of Bim enhanced the apoptotic threshold [30]. In the absence of stress, Bim can be isolated by dynein light chain 1 in microtubules. When cells are subjected to pro-apoptotic stimulation, Bim is delivered from the microtubules, triggering mitochondria- related apoptosis initiation [31].
In the current work, we showed that change of miR-30e-5p expression was conversely related to the expression of Bim. Moreover, miR-30e-5p overexpression ameliorated cell apoptosis and Caspase-3 initiation in hiPSC-CMs exposed to hypoxia. Therefore, we showed that miR-30e-5p enhanced cell survival by posttranscriptionally regulating Bim. Interestingly, beyond its apoptotic effect, a recent study showed that Bim can also control autophagy via interacting with Beclin-1 and further inhibit autophagy initiation through mislocalizing Beclin-1 to dynein light chain 1[32].
Apoptosis and autophagy are largely known as two interactive stress-response mechanisms that affect cell metabolism [33]. The interaction between apoptosis and autophagy is complicated and extremely context-dependent. Apart from the rare conditions under which autophagy initiates apoptosis, autophagy may deter the initiation of apoptosis or enhance the threshold of apoptosis by degrading dysfunctional mitochondria and adverse proteins[34]. Inversely, activated Caspase-3 instantly eradicates key autophagy components, such as p62 and Beclin-1, thus halting the process of autophagy [35, 36]. In addition, Beclin-1 fragments are locally restricted in the mitochondria and thus accelerate apoptosis[36]. It comes to light that the response to hypoxia differs in various hypoxic gradients and different cell types. Controlled hypoxia conditions can provoke autophagy initiation to promote cell survival[37]. Conversely, heavy hypoxia may curb autophagy activation and cause cell death[38].
Our study revealed significant autophagy suppression with a decrease in Beclin-1 and p62 in hiPSC-CMs after 24 h hypoxia treatment. miR-30e-5p overexpression effectively repaired autophagy, evidenced by the expression p62 and Beclin-1 in hiPSC-CMs exposed to 24 h of hypoxia. This autophagy restoration may be resolved by posttranscriptional modulation of Bim via miR-30e-5p. Therefore, autophagy might protect against hypoxia-induced apoptosis. In a word, our results demonstrate that miR-30e-5p may protect against hypoxia-induced hiPSC-CM apoptosis targeting of Bim.
Although our work demonstrates that miR-30e-5p may protect against hypoxia-induced hiPSC-CM apoptosis via inhibition of Bim, it is important to note that there are some limitations to our work. First, additional reported, experimentally verified targets, such as TP53, HSPA5, and BNIP3, are related to controlling cell apoptosis. Other reported miR-30e-5p targets might also govern cell survival modulation. Given other targets of miR-30e-5p reported might also modulate Bim and autophagy, the potential of indirect regulation by these proteins should be fully investigated. Second, considering that the miRNA-target interaction might change among different cell types, the miR-30e-5p--Bim interaction confirmed in HEK 293 cells in our finding might not act at the same level in hiPSC-CMs. Third, the mechanism underlying miR-30e-5p overexpression in response to hypoxia-induced hiPSC-CM apoptosis remains to be clarified. Further efforts are required to elucidate whether miR-30e-5p overexpression can decrease myocardial infarct size and enhance ventricular function after CME in vivo. In spite of these limitations, our results demonstrate that miR-30e-5p may protect against hypoxia-induced hiPSC-CM apoptosis through targeting of Bim. Further efforts showed that this effect was fulfilled at least partially via posttranscriptional regulation of Bim and initiation of autophagy. These results may identify a potential therapeutic target for CME.
Supplementary Material
Movie S1.
Movie S2.
Acknowledgements
This work was supported by National Natural Science Foundation of China (grant no.81770346) and Appropriate Technology for Health Care Research and Development Projects of Guangxi, China (grant no. S2015 21).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Erbel R, Heusch G. Coronary microembolization. Journal of the American College of Cardiology. 2000;36:22-4
2. Galiuto L, Garramone B, Scara A, Rebuzzi AG, Crea F, La Torre G. et al. The extent of microvascular damage during myocardial contrast echocardiography is superior to other known indexes of post-infarct reperfusion in predicting left ventricular remodeling: results of the multicenter AMICI study. Journal of the American College of Cardiology. 2008;51:552-9
3. Wu X, Mintz GS, Xu K, Lansky AJ, Witzenbichler B, Guagliumi G. et al. The relationship between attenuated plaque identified by intravascular ultrasound and no-reflow after stenting in acute myocardial infarction: the HORIZONS-AMI (Harmonizing Outcomes With Revascularization and Stents in Acute Myocardial Infarction) trial. JACC Cardiovascular interventions. 2011;4:495-502
4. Gao YH, Qian JY, Chen ZW, Fu MQ, Xu JF, Xia Y. et al. Suppression of Bim by microRNA-19a may protect cardiomyocytes against hypoxia-induced cell death via autophagy activation. Toxicology letters. 2016;257:72-83
5. Su M, Wang J, Wang C, Wang X, Dong W, Qiu W. et al. MicroRNA-221 inhibits autophagy and promotes heart failure by modulating the p27/CDK2/mTOR axis. Cell death and differentiation. 2015;22:986-99
6. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-97
7. De Rosa S, Indolfi C. Circulating microRNAs as Biomarkers in Cardiovascular Diseases. Exs. 2015;106:139-49
8. Gladka MM, van Rooij E. AntimiR-34a to Enhance Cardiac Repair After Ischemic Injury. Circulation research. 2015;117:395-7
9. Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S. et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nature communications. 2012;3:1078
10. Gedik N, Thielmann M, Kottenberg E, Peters J, Jakob H, Heusch G. et al. No evidence for activated autophagy in left ventricular myocardium at early reperfusion with protection by remote ischemic preconditioning in patients undergoing coronary artery bypass grafting. PloS one. 2014;9:e96567
11. Li D, Wang J, Hou J, Fu J, Liu J, Lin R. Salvianolic acid B induced upregulation of miR-30a protects cardiac myocytes from ischemia/reperfusion injury. BMC complementary and alternative medicine. 2016;16:336
12. Yang Y, Li Y, Chen X, Cheng X, Liao Y, Yu X. Exosomal transfer of miR-30a between cardiomyocytes regulates autophagy after hypoxia. Journal of molecular medicine (Berlin, Germany). 2016;94:711-24
13. Sinnecker D, Laugwitz KL, Moretti A. Induced pluripotent stem cell-derived cardiomyocytes for drug development and toxicity testing. Pharmacology & therapeutics. 2014;143:246-52
14. Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, Wang L. et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell stem cell. 2013;12:101-13
15. Bhattacharya S, Burridge PW, Kropp EM, Chuppa SL, Kwok WM, Wu JC. et al. High efficiency differentiation of human pluripotent stem cells to cardiomyocytes and characterization by flow cytometry. Journal of visualized experiments: JoVE. 2014:52010
16. Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM. et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E1848-57
17. Heusch G, Kleinbongard P, Bose D, Levkau B, Haude M, Schulz R. et al. Coronary microembolization: from bedside to bench and back to bedside. Circulation. 2009;120:1822-36
18. Li XD, Yang YJ, Geng YJ, Cheng YT, Zhang HT, Zhao JL. et al. The cardioprotection of simvastatin in reperfused swine hearts relates to the inhibition of myocardial edema by modulating aquaporins via the PKA pathway. International journal of cardiology. 2013;167:2657-66
19. Wang XT, Wu XD, Lu YX, Sun YH, Zhu HH, Liang JB. et al. Potential Involvement of MiR-30e-3p in Myocardial Injury Induced by Coronary Microembolization via Autophagy Activation. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2017;44:1995-2004
20. Liang J, Li L, Sun Y, He W, Wang X, Su Q. The protective effect of activating Nrf2 / HO-1 signaling pathway on cardiomyocyte apoptosis after coronary microembolization in rats. BMC cardiovascular disorders. 2017;17:272
21. Li J, Donath S, Li Y, Qin D, Prabhakar BS, Li P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS genetics. 2010;6:e1000795
22. Zhang T, Tian F, Wang J, Jing J, Zhou SS, Chen YD. Endothelial Cell Autophagy in Atherosclerosis is Regulated by miR-30-Mediated Translational Control of ATG6. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2015;37:1369-78
23. Chen M, Ma G, Yue Y, Wei Y, Li Q, Tong Z. et al. Downregulation of the miR-30 family microRNAs contributes to endoplasmic reticulum stress in cardiac muscle and vascular smooth muscle cells. International journal of cardiology. 2014;173:65-73
24. Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I. et al. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circulation research. 2009;104:170-8 6p following 8
25. Saddic LA, Muehlschlegel JD. Sarco"MiR" friend or foe: a perspective on the mechanisms of doxorubicin-induced cardiomyopathy. Annals of translational medicine. 2016;4:203
26. Shen Y, Shen Z, Miao L, Xin X, Lin S, Zhu Y. et al. miRNA-30 family inhibition protects against cardiac ischemic injury by regulating cystathionine-gamma-lyase expression. Antioxidants & redox signaling. 2015;22:224-40
27. Ajabnoor GM, Crook T, Coley HM. Paclitaxel resistance is associated with switch from apoptotic to autophagic cell death in MCF-7 breast cancer cells. Cell death & disease. 2012;3:e260
28. Rahmani M, Aust MM, Attkisson E, Williams DC Jr, Ferreira-Gonzalez A, Grant S. Inhibition of Bcl-2 antiapoptotic members by obatoclax potently enhances sorafenib-induced apoptosis in human myeloid leukemia cells through a Bim-dependent process. Blood. 2012;119:6089-98
29. Moldoveanu T, Follis AV, Kriwacki RW, Green DR. Many players in BCL-2 family affairs. Trends in biochemical sciences. 2014;39:101-11
30. Pernaute B, Spruce T, Smith KM, Sanchez-Nieto JM, Manzanares M, Cobb B. et al. MicroRNAs control the apoptotic threshold in primed pluripotent stem cells through regulation of BIM. Genes & development. 2014;28:1873-8
31. Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:2432-7
32. Luo S, Garcia-Arencibia M, Zhao R, Puri C, Toh PP, Sadiq O. et al. Bim inhibits autophagy by recruiting Beclin 1 to microtubules. Molecular cell. 2012;47:359-70
33. Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nature reviews Molecular cell biology. 2014;15:81-94
34. Mukhopadhyay S, Panda PK, Sinha N, Das DN, Bhutia SK. Autophagy and apoptosis: where do they meet? Apoptosis: an international journal on programmed cell death. 2014;19:555-66
35. Norman JM, Cohen GM, Bampton ET. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy. 2010;6:1042-56
36. Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I. et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell death & disease. 2010;1:e18
37. Mazure NM, Pouyssegur J. Hypoxia-induced autophagy: cell death or cell survival? Current opinion in cell biology. 2010;22:177-80
38. Yan L, Yang H, Li Y, Duan H, Wu J, Qian P. et al. Regulator of calcineurin 1-1L protects cardiomyocytes against hypoxia-induced apoptosis via mitophagy. Journal of cardiovascular pharmacology. 2014;64:310-7
Author contact
![]() Corresponding author: Lang Li MD, PhD, Professor, Department of Cardiology, The First Affiliated Hospital of Guangxi Medical University, No.6 Shuangyong Road, Nanning 530021 Guangxi, People's Republic of China, Phone: +86-0771-5331171; Fax: +86-0771-5331171; Email: drlilangcom
Corresponding author: Lang Li MD, PhD, Professor, Department of Cardiology, The First Affiliated Hospital of Guangxi Medical University, No.6 Shuangyong Road, Nanning 530021 Guangxi, People's Republic of China, Phone: +86-0771-5331171; Fax: +86-0771-5331171; Email: drlilangcom