Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(6):3108-3126. doi:10.7150/ijbs.128409 This issue Cite
Research Paper
FAM177A1 disrupts SIRT3-SOD2 signaling to drive mitochondrial dysfunction-mediated VSMC phenotypic switching in vascular remodeling
Ruiqi Mao1,2,3#, Yi Guo4#, Ling Jiang5#, Shichen Bu6, Ming Chen7, Minglu Liang2,3, Xinyuan Gao1,2,3, Yichen Wu1,2,3, Wenjing Xu1,2,3, Zilong Chen2,3, Kai Huang2,3,8 ![]() , Xiaoguang Li2,3
, Xiaoguang Li2,3 ![]() , Cheng Wang2,3,9
, Cheng Wang2,3,9 ![]()
1. Department of Cardiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
2. Hubei Key Laboratory of Metabolic Abnormalities and Vascular Aging, Huazhong University of Science and Technology, Wuhan, China.
3. Hubei Clinical Research Center for Metabolic and Cardiovascular Disease, Huazhong University of Science and Technology, Wuhan, China.
4. Cardiovascular Center, Liyuan Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
5. Department of Geriatrics, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
6. Department of Cardiology, Xiamen Cardiovascular Hospital of Xiamen University, School of Medicine, Xiamen University, Xiamen, Fujian, China.
7. Department of Cardiovascular Surgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
8. State Key Laboratory of Metabolism and Regulation in Complex Organisms, Taikang Center for Life and Medical Sciences, School of Basic Medical Sciences, Wuhan University, Wuhan, China.
9. Department of Rheumatology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
#Equal contribution.
Received 2025-11-5; Accepted 2026-2-6; Published 2026-2-26
Abstract

Aims: Vascular remodeling involves structural and functional vascular changes in response to injury, aging, and disease. A key pathological feature is vascular smooth muscle cells (VSMCs) phenotypic switching, which is accompanied by mitochondrial dysregulation. Metabolic reprogramming resembling the Warburg effect alongside mitochondrial oxidative damage collectively drive this pathological VSMC transdifferentiation. We hypothesized that targeting mitochondrial ROS could restore mitochondrial integrity and enhance oxidative phosphorylation (OXPHOS) to counteract both oxidative damage and metabolic reprogramming in cardiovascular diseases associated with vascular remodeling. We proposed that the uncharacterized membrane-associated protein FAM177A1 drives VSMC mitochondrial oxidative impairment and metabolic reprogramming, thereby promoting VSMC phenotypic switching and vascular dysfunction.
Methods and Results: We modeled vascular remodeling using global Fam177a1 knockout rats subjected to carotid balloon injury, VSMC-specific AAV-mediated Fam177a1 knockdown in carotid artery ligation mice, and using ApoE-/- mice fed a 12-week high-fat diet to induce atherosclerosis; in vitro VSMCs with platelet-derived growth factor-bb (PDGF-BB) stimulation further elucidated FAM177A1's role in phenotypic switching. FAM177A1 expression was significantly elevated in injured and atherosclerotic aortas, while its deficiency suppressed neointimal hyperplasia and atherosclerosis development. FAM177A1 deficiency upregulated mitochondrial functional genes, enhanced mtDNA biogenesis, reduced ROS accumulation, maintained redox homeostasis, and preserved mitochondrial membrane potential (ΔΨm). Moreover, FAM177A1 deficiency enhanced oxidative phosphorylation (OXPHOS) while reducing glycolytic flux, thereby improving bioenergetic efficiency and promoting a contractile phenotype. Molecular analysis revealed that FAM177A1 disrupted SIRT3-SOD2 binding, leading to elevated SOD2 K68 acetylation which decreased SOD2 activity and stability. Under pathological condition, this dysregulated cascade increased mitochondrial ROS, impaired mitochondrial function, thereby accelerating VSMC phenotypic switching.
Conclusion: We identify FAM177A1 as a key mitochondrial regulator that drives VSMC switching through SIRT3-SOD2 axis disruption. Targeting FAM177A1 restores redox-metabolic homeostasis through scavenging ROS and improving OXPHOS, establishing it as a novel therapeutic target against vascular remodeling.
Introduction
Cardiovascular diseases (CVD), primarily stroke and ischemic heart disease, are the leading causes of global disability and mortality[1, 2]. Furthermore, the worldwide burden of CVD, driven by multiple risk factors, significantly contributes to rising healthcare costs[3, 4]. Reducing CVD incidence, premature disability, and the resultant increase in public health spending is therefore a critical priority for national health systems and researchers. Chronic vascular pathologies—including atherosclerosis, post-angioplasty restenosis, aortic aneurysm, and hypertension—are consistently characterized by vascular remodeling[5-7]. Consequently, targeting vascular smooth muscle cell (VSMC) phenotypic switching represents a promising strategy for preventing and treating these remodeling-associated diseases[8].
Beyond well-studied growth factors like transforming growth factor (TGF-β)[9], fibroblast growth factor (FGF)[10] and platelet-derived growth factor BB (PDGF-BB)[11], reactive oxygen species (ROS) emerge as critical factor regulating VSMC phenotypic switching[12]. While basal ROS levels are essential for vascular homeostasis[13], dysregulation of ROS-generating or scavenging enzymes induces oxidative stress, driving pathological VSMC transdifferentiation and proliferation[14]. Elevated ROS levels are associated with atherosclerosis, intimal injury, and diabetes. Furthermore, PDGF-BB or bradykinin stimulation further increases VSMC ROS[15]. Crucially, ROS promote VSMC proliferation, migration, and altered differentiation marker expression—core hallmarks of phenotypic switching[16]. ROS also facilitate PDGF-BB- or angiotensin II (Ang-II)-induced switching and vascular remodeling[17]. In atherosclerosis, ROS promote the synthetic VSMC phenotype via the ELK-1/SRF pathway[18], while under inflammation, ROS activate NF-κB, upregulating osteopontin and suppressing myocardin-dependent contractile markers like SM22[19]. Thus, oxidative stress is fundamentally implicated in VSMC switching during vascular remodeling.
Mitochondria serve as a major ROS source yet are highly susceptible to oxidative damage, frequently becoming dysfunctional in CVD[20]. During VSMC switching, mitochondria shift metabolism from oxidative phosphorylation (OXPHOS) to glycolysis[21]. This Warburg-like adaptation supports the heightened bioenergetic and biosynthetic demands of the proliferative/migratory state, facilitating switching and remodeling[22, 23]. Enhancing OXPHOS to suppress this reprogramming is thus a promising therapeutic strategy[22, 24]. However, pathological mitochondrial ROS (mtROS) often compromise VSMC mitophagy and mitochondrial OXPHOS[25]. Consequently, metabolic reprogramming and mitochondrial dysfunction frequently coexist during switching. Superoxide dismutase 2 (SOD2), the primary mitochondrial superoxide scavenger[26], constitutes a key regulatory node. Nevertheless, whether targeting SOD2 can effectively break the vicious cycle by scavenging mtROS, restoring OXPHOS, and preventing pathological VSMC switching remains a fundamental question.
FAM177A1 is an evolutionarily conserved (human to zebrafish) membrane-associated protein, primarily Golgi-localized[27, 28]. Emerging evidence has implicated FAM177A1 in various pathological conditions, including neurodevelopmental disorders[28], inflammatory diseases[29-31], and cancer[32]. Biallelic FAM177A1 variants cause a defined neurodevelopmental disorder featuring macrocephaly, global developmental delay, intellectual disability, seizures, behavioral abnormalities, hypotonia, and gait disturbances in five individuals across three families[28]. FAM177A1 deficiency dysregulates pathways involving apoptosis, inflammation, and proliferation suppression in human fibroblasts and zebrafish larvae[33]. Furthermore, FAM177A1 critically regulates innate immune responses, including TRAF6-NF-κB and Toll-like receptor 4 (TLR4) signaling[29, 30]. Reduced expression correlates with decreased primary biliary cholangitis incidence[31]. Within the Golgi, FAM177A1 interacts with VPS13B, influencing lipid trafficking and membrane dynamics[27]. While these findings existed linking FAM177A1 to neurodevelopmental disorders, macrophage-mediated inflammation, and potential associations with proliferation, no study has investigated the connection between FAM177A1 and vascular remodeling. It's function and related molecular mechanisms in VSMC switching remain undefined. In this study, we revealed that FAM177A1 is a critical regulator of VSMC phenotypic switching and promotes atherosclerosis and neointimal hyperplasia. Mechanistically, FAM177A1 was found to regulate the cellular fate of VSMCs by disrupting mitochondrial redox homeostasis. We serendipitously discovered its mitochondrial localization, revealing a key role for this pool in modulating the mitochondrial SIRT3-SOD2-mtROS axis and mitochondrial bioenergetics. Thus, targeting FAM177A1 may represent a promising therapeutic strategy for intervening in vascular remodeling.
Methods
Animal experiments, anesthesia and euthanasia
Fam177a1 knockout rats were constructed by Wuhan Cyagen Biotechnology. 20bp (AAGAAAGATGTGCTGCCCAC) were deleted on exon 2 of Fam177a1 gene. ApoE-/- and C57BL/6J mice were purchased from Vital River Laboratories. Male Sprague-Dawley rats were purchased from Wuhan Shubeili biotechnology. For the mouse carotid artery ligation and rat carotid artery balloon injury models, anesthesia was induced with 100% O2 / 4% isoflurane and maintained throughout the procedure using 100% O₂ / 2% isoflurane. At the end of the experiments, all mice were euthanized under deep anesthesia with 100% O₂ / 5% isoflurane, followed by tissue collection. All mice and rats were housed in specific pathogen-free (SPF) facilities at Huazhong University of Science and Technology under a 12-hour light/dark cycle, with free access to food and water. All animal experiments were conducted with permission the Animal Experimentation Committee of Huazhong University of Science and Technology (IACUC No. 4635), in compliance with the Animals (Scientific Procedures) Act of 1986 (United Kingdom), Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes, and the NIH Guide for the Care and Use of Laboratory Animals.
Establishment of mouse carotid artery ligation model
Eight weeks old male C57BL/6J mice were injected the AAV-EnSM22a-shRNA-Fam177a1. Carotid artery ligation after anesthesia as previously described[34] were subjected after 3 weeks. Two weeks after surgery, carotid arteries were collected, serially sectioned, and stained.
Establishment of rat ballon injury model
Male Sprague-Dawley rats (210-230 g) and 2.0-French catheter were used for the carotid artery injury model. The surgical procedure was performed as previously described[33]. The contralateral untreated carotid artery was used as control. Carotid arteries were harvested at predetermined experimental time points, followed by serial sectioning and staining. For 3-TYP treatments, rats were randomly grouped after ballon-catheter injury and the 3-TYP group rats were injected of 3-TYP (50mg/kg/d) intraperitoneally once a day.
Establishment of atherosclerosis model
Eight-week-old ApoE-/- mice were fed with high-fat diet (D12108C, Medicience, Jiangsu, China) for 12 weeks. Mice were sacrificed after anesthesia to collect the blood, heart and aortas. The collected blood was centrifuged after overnight standing, and the supernatant was used for blood lipid profiling. The intact aorta was stained with Oil Red O and photographed (Servicebio). The heart was cryosectioned, with serial sections of the aortic sinus processed for HE, Masson, and Oil Red O staining.
Results
FAM177A1 is upregulated in VSMC during phenotypic switching
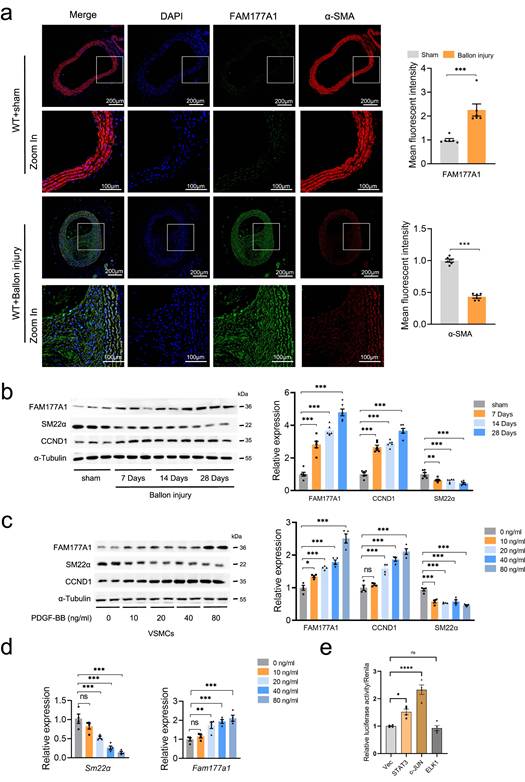
To investigate the connection between FAM177A1 and VSMCs, published single-cell RNA sequencing (scRNA-seq) data (GSE174098) derived from left (control) and right (balloon injury) carotid arteries of rats were applied to analyze Fam177a1 dynamic in vascular injury stenosis. A t-SNE dimension reduction plot of 7 cell types showed the proportions of different cell types, including endothelial cells (ECs), VSMCs, fibroblasts (FB), macrophages and unknown cells (Fig. S1a). Through vascular remodeling, VSMC phenotype switched to fibroblast phenotype, accompanied by loss of contractile VSMCs markers and elevation of cell proliferation, migration and ECM remodeling markers[35, 36]. Supportingly, levels of SMC-specific marker genes (Acta2, Tagln, Cnn1) were declined in FB cell group (Fig. S1b-c). Noteworthily, the Fam177a1 expression was observed tendency towards higher in FB compared to VSMC (Fig. S1c). We next examined the expression of FAM177A1 by constructing rat carotid balloon injury. Rats were sacrificed 0/7/14/28 days after surgery and carotid artery specimens were collected from the sham group (Sham) and the surgical group (Injury). Immunofluorescence assay showed that FAM177A1 was significantly upregulated in the hyperplastic intima of rat carotid arteries 28 days after balloon injury, accompanied with decreased α-SMA (VSMCs identity marker) (Fig. 1a). Moreover, western blots showed time-dependent upregulation of FAM177A1 expression with a concomitant reduction in SM22α (contractile marker) and elevation in cyclin D1 (CCND1, proliferation marker) in extraction of carotid artery from carotid ballon injury rats, compared with sham group (Fig. 1b). Next, we performed in vitro experiments that quiescent-state primary VSMCs were stimulated with PDGF-BB to induce VSMC phenotypic switching[37, 38]. Consistently, we observed PDGF-BB concentration dependent dynamic of decreased SM22α and increased CCND1 (Fig. 1c), indicating a contractile quiescent phenotype switching to a synthetic one. Importantly, the protein expression of FAM177A1 was increasing upon the increased PDGF-BB stimulation (Fig. 1c). Along this vein, we found that Fam177a1 mRNA levels were significantly elevated in a PDGF-BB concentration-dependent manner as well (Fig. 1d). Based on the observation that FAM177A1 expression changed at the RNA level following PDGF-BB stimulation, we further employed luciferase reporter assays to investigate which specific transcription factors or signaling pathways control its expression under PDGF-BB stimulation. Transfection with STAT3 or c-JUN plasmids promoted the luciferase activity/renilla ratio, thereby enhancing FAM177A1 transcription, whereas transfection with ELK1 plasmid did not significantly promote transcription. Collectively, these data suggested that both c-Jun/AP-1 and STAT3 are potential transcription factors mediating the PDGF-BB-induced transcriptional upregulation of FAM177A1(Fig. 1e).
FAM177A1 upregulation correlated with pathological vascular remodeling. a. Representative immunofluorescence images and quantification of α-SMA (red) and FAM177A1 (green) in carotid arteries of WT+Ballon injury group (n = 6) and sham-operated group (WT+sham) (n = 6) post 28 days. Scale bar: 200 μm and 100 μm (zoom in). b. Representative Western blotting and quantification of FAM177A1, SM22α and CCND1 in the rat carotid arteries of sham-operated and post-ballon injury at different time intervals (n = 6). c. Representative Western blotting and quantification of CCND1, SM22α and Fam177a1 in rat VSMCs under different PDGF-BB concentrations (n = 3). d. qPCR analysis of the relative mRNA level of Sm22α and Fam177a1 in rat VSMCs under different concentrate PDGF-BB stimulation (n = 3). 18S was used for control. e. Dual-luciferase reporter assays performed to measure Fam177a1 promoter activity in A7r5 cells transfected with empty vector (Vec) or STAT3, c-JUN, and ELK1 overexpression plasmids for 48 hours (n = 3). Vec was used for control. All Data are presented as means ± SEM. P-values are calculated using the Student's t-test (unpaired) in a and one-way ANOVA with a post hoc test of Tukey's analysis in b-e,. ns, p>0.05, *p <0.05, **p<0.01, ***p<0.001. SM22α, smooth muscle 22 alpha; CCND1, cyclin D1; PDGF-BB, platelet-derived growth factor-bb.
Additionally, we established a conventional atherosclerotic (AS) model using ApoE-/- mice induced with a 12-week high-fat diet (HFD). We observed that FAM177A1 expression was upregulated in VSMCs within atherosclerotic plaques, concomitant with a decline in α-SMA expression (Fig. S1d). These findings suggest that FAM177A1 is upregulated during HFD-induced VSMC phenotypic transformation in mice. Humans express two FAM177A1 isoforms (isoform 1 and 2), resulting from alternative initiation sites that generate distinct N-terminal regions due to the exclusion or inclusion of a 21-amino acid fragment[39]. Notably, humans also produce a secreted form containing an N-terminal 21-amino acid signal peptide (the signal peptide was predicted by SignalP6.0), detectable in human blood[40]. To investigate the clinical relevance of FAM177A1 in AS, we collected serum samples and clinical records from 99 individuals undergoing routine health examinations (Table S2). Based on coronary angiography (stenosis ≥50%) and carotid ultrasound (presence of plaque or intima-media thickness ≥1.0 mm) criteria, 45 subjects (37 male, 8 female) were categorized into the AS group. The remaining 54 subjects (44 male, 10 female) served as healthy controls. ELISA analysis revealed significantly elevated serum FAM177A1 levels in AS patients (189.2 ± 33.47 pg/ml) compared to controls (167.6 ± 35.54 pg/ml; p = 0.0025) (Fig. S1e). Its elevated serum levels in AS patients demonstrate considerable translational potential as a diagnostic biomarker for atherosclerosis.
In summary, these findings strongly suggested that FAM177A1 is upregulated in VSMC during phenotypic switching, and suggested that it may play a crucial role in vascular remodeling and maintaining the contractile phenotype of VSMCs.
Fam177a1 knockout suppresses vascular remodeling in balloon injury-induced neointimal hyperplasia and atherosclerosis
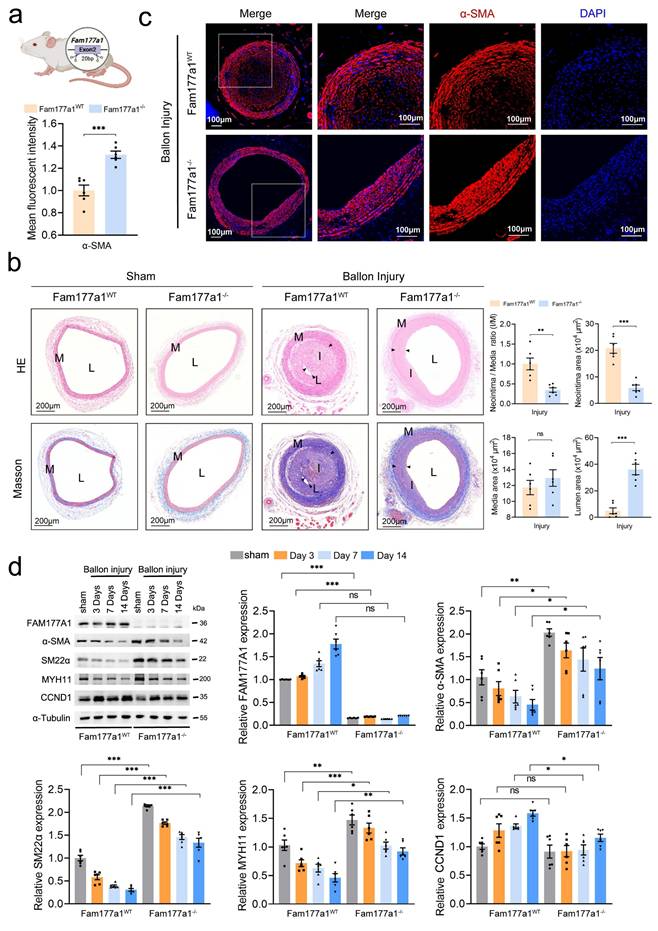
To further elucidate the function of FAM177A1 in vascular remodeling, we generated Fam177a1 knockout (Fam177a1-/-) rats through a 20-bp deletion in exon 2 of the gene (Fig. 2a). Next, we used 1.5mm balloon catheter to construct the carotid artery neointima injury model on Fam177a1-/- and WT (Fam177a1WT) rats. H&E and Masson staining revealed significant compensatory dilatation in these intact arteries, which likely developed in response to luminal stenosis caused by neointimal hyperplasia in the operated vessels (Fig. 2b). Moreover, genetic ablation of Fam177a1 suppressed postinjury neointima formation (20.79±4.536 x104μm2 vs 5.88±2.581 x104μm2, p<0.0001) and collagen deposition (Fig. 2b). Compared to Fam177a1WT rat, Fam177a1-/- rat exhibited a significantly larger lumen area (4.963±5.387 x104μm2 vs 36.08±9.552 x104μm2, p<0.0001) (Fig. 2b). These data indicated that Fam177a1 knockout suppresses vascular remodeling in balloon injury-induced neointimal hyperplasia. Additionally, immunofluorescence assays revealed that carotid arteries from Fam177a1-/- rat exhibited higher levels of α-SMA protein compared with those from Fam177a1WT rat following injury (Fig. 2c), indicating enhanced contractile function. Western blots analysis of injured vessels demonstrated that FAM177A1 deficiency upregulated VSMC contractile proteins (α-SMA, SM22α, MYH11) and downregulated the proliferation marker CCND1 at sequential time points post-injury (0, 3, 7, and 14 days) (Fig. 2d).
Fam177a1 knockout alleviated vascular intimal hyperplasia in vivo. a. Schematic illustration of the generation of Fam177a1 knockout rat. b. Representative immunofluorescence images and quantification of α-SMA (red) in male Fam177a1WT and Fam177a1-/- rats carotid arteries post ballon injury 28 days (n = 6). DAPI was used for nuclear staining (blue). Scale bar: 100μm. Statistical analysis of αSMA mean fluorescence intensity (total intensity/area) were shown on the left (normalized to Fam177a1WT group). c. Left, Representative cross sections of H&E and Masson in male Fam177a1WT and Fam177a1-/- rats carotid arteries post ballon injury 28 days. Scale bar=200μm. Right, quantitative analysis of the neointima/media ratio(I/M), neointima areas, media areas and lumen areas in HE-stained sections from male Fam177a1WT and Fam177a1-/- rats carotid arteries post ballon injury 28 days (n = 6). d. Representative Western blotting and quantification in the Fam177a1WT and Fam177a1-/- rats carotid arteries of sham-operated or post balloon injury at different time intervals groups (n = 6). α-Tubulin was used for control. Data were presented as relative fold change to Fam177a1WT sham group. All Data are presented as means ± SEM. P-values are calculated using the Student's t-test (unpaired) in b and c, and one-way ANOVA with a post hoc test of Tukey's analysis in d. *p <0.05, **p<0.01, ***p<0.001. H&E, Hematoxylin and Eosin; α-SMA, α smooth muscle actin; MYH11, myosin heavy chain 11.
In parallel, we constructed recombinant AAV9 vectors carrying either a short hairpin RNA targeting mouse Fam177a1 (AAV-EnSM22α-shFam177a1, targeting sequence: GCAGAUAAGUAAUGAAAGA) or a scramble shRNA control (AAV-EnSM22α-shScr), both driven by the vascular smooth muscle-specific SM22α promoter. Three weeks after tail vein injection of these recombinant AAVs, western blot analysis confirmed the successful knockdown of FAM177A1 in the aorta (Fig. S2a). Next, we utilized these recombinant AAVs in a conventional mouse carotid artery injury model in which the left common carotid artery was ligated with a 6-0 silk suture (Fig. S2b). Consistent with our previous results, H&E staining revealed that wire injury-induced neointima formation was attenuated in the carotid arteries of mice injected with AAV-SM22α-shFam177a1 (Fig. S2c). These findings demonstrate a prominent regulatory role for FAM177A1 in neointima formation following carotid artery ligation.
To investigate the function of FAM177A1 in atherosclerosis-associated vascular remodeling, ApoE-/- mice fed with HFD for 12 weeks were injected with either AAV-EnSM22α-shFam177a1 or AAV-EnSM22α-Scr (Fig. 3a). No significant differences were observed in liver Oil Red O staining or blood lipid levels between the shFam177a1 and shScr groups (Fig. S2d-e), indicating comparable systemic metabolic profiles. En-face Oil Red O staining of the entire aorta revealed that Fam177a1 knockdown in VSMCs via AAV-mediated SM22α promoter-driven shRNA delivery significantly reduced plaque area (Fig. 3b). Analysis of Oil Red O-stained aortic roots further demonstrated reduced plaque area in the shFam177a1 group (0.4899 ± 0.09239 mm² vs. 0.7590 ± 0.1433 mm², p=0.0031) (Fig. 3c, d). Furthermore, H&E and Masson staining of aortic roots showed that Fam177a1 knockdown resulted in a smaller necrotic core area (0.07944 ± 0.03022 mm² vs. 0.1691 ± 0.04373 mm², p=0.0020) and altered collagen content within the lesions (Fig. 3c, d). These findings suggest that Fam177a1 knockdown reduces atherosclerotic plaque burden and promotes plaque stability.
Fam177a1 specific knockdown in VSMCs mitigated atherosclerosis in vivo. a. Experimental scheme. ApoE-/- mice were injected with AAV-EnSM22α-shRNA-Scramble or AAV-EnSM22α-shRNA-Fam177a1 via the tail vein, followed by a 12-week high-fat diet to establish the atherosclerosis model with smooth muscle-specific knockout of Fam177a1. Blood, aortas, and heart were collected for further analyses. b. Representative image of Oil Red O-stained whole aortas from mice as indicated (n= 12). The relative En face atherosclerotic plaque area was quantified compared to AAV-EnSM22α-shRNA-Scramble group. Scale bar: 0.5mm. c. Representative image of cross section of the H&E, Oil red O and Masson-stained aortic roots from mice as indicated. Scale bar: 0.5mm and 0.2mm (zoom in). d. Quantitative analysis of plaque areas, plaque/aortic area ratio and necrotic areas in H&E, Oil red O and Masson-stained aortic roots from mice as indicated (n = 6). Data were compared to AAV-EnSM22α-shRNA-Scramble group. e. Representative immunofluorescence images of CD68(green) and αSMA (red) on cross section of the aortic roots of indicated mice (n = 6). DAPI was used for nuclear staining(blue). Scale bar: 100μm. f. Statistical analysis of CD68 and αSMA mean fluorescence intensity. All Data are presented as means ± SEM. P-values are calculated using the Student's t-test (unpaired) in b, d and f. *p <0.05, **p<0.01, ***p<0.001. Scr, Scramble.
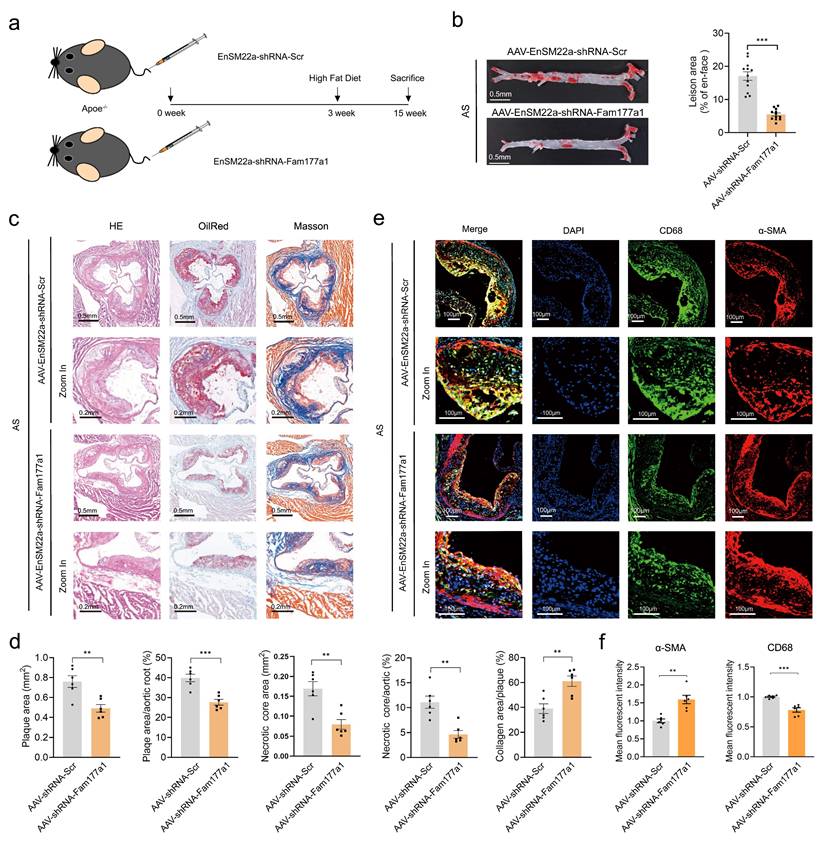
Given that plaque rupture is inversely correlated with contractile VSMC abundance and positively correlated with VSMC-derived macrophage-like cells[41-44], we assessed VSMC and macrophage markers via immunofluorescence. The aortic root lesions of the AAV-SM22α-shFam177a1 group exhibited higher levels of α-SMA (VSMCs marker) and lower levels of CD68 (macrophage marker) compared to controls (Fig. 3e, f). Collectively, these results indicate that FAM177A1 regulates atherosclerosis progression and plaque stability, potentially by modulating VSMC phenotype and function.
FAM177A1 promotes VSMC phenotype switching from the contractile to synthetic phenotype
Given that FAM177A1 play a vital role in atherosclerosis and neointimal hyperplasia in in the tunica media, we next conducted in vitro cellular experiments to investigate the effect of FAM177A1 on VSMC phenotype. At quiescent state without PDGF-BB stimulation, immunofluorescence staining of F-actin revealed that Fam177a1 knockout preserved the spindle-like morphology of rat primary VSMCs, preventing a shift toward large, polygonal shapes observed in controls (Fig.S3a). Conversely, FAM177A1 overexpression induced a marked transition from spindle-shaped to polygonal morphology (Fig.S3d), indicating a synthetic phenotype. Fam177a1-/- VSMCs exhibited upregulated contractile proteins (α-SMA, MYH11, SM22α, CNN1) and downregulated proliferation markers (PCNA, CCND1) at both mRNA and protein levels (Fig.S3b, c). In contrast, FAM177A1-overexpressing VSMCs showed attenuated contractile protein expression and elevated proliferation markers (Fig.S3e, f). These results demonstrate that FAM177A1 promotes cell-autonomous VSMC phenotype switching.
FAM177A1 exacerbates PDGF-BB-induced VSMC phenotypic switching
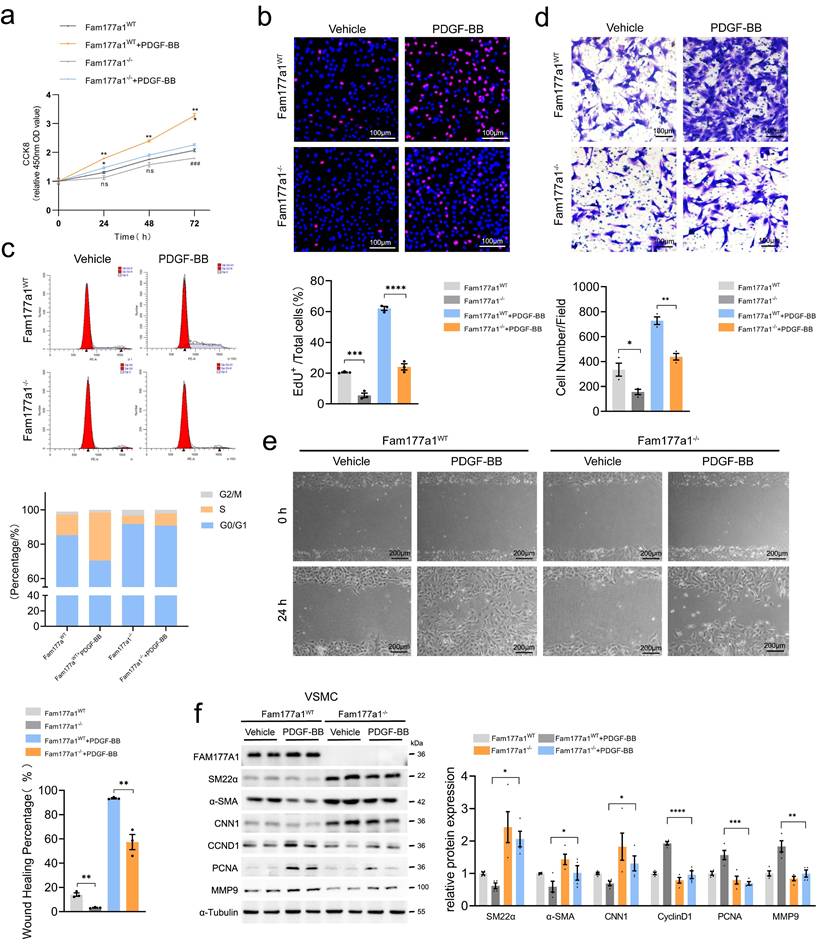
To further explore the regulation of VSMC phenotype by FAM177A1 under pathological conditions, we treated VSMCs with PDGF-BB (20 ng/ml) to simulate a disease-relevant microenvironment. Under PDGF-BB stimulation mimicking pathological conditions, Fam177a1 knockout significantly decreased CCK-8 activity, EdU+ cell proportion, and S-phase fraction in primary VSMCs compared to Fam177a1WT (Fig. 4a-c), indicating suppressed proliferation. Transwell and wound healing assays confirmed that Fam177a1 knockout inhibited PDGF-BB-induced VSMC migration (Fig. 4d-e). Consistent with this, Fam177a1-/- VSMCs displayed elevated contractile proteins (SM22α, α-SMA, CNN1), and suppressed proliferation (Cyclin D1, PCNA) and migration markers (MMP9) (Fig. 4f). Conversely, FAM177A1 overexpression exacerbated PDGF-BB-induced VSMC proliferation, migration, and contractile-synthetic protein shifts (Fig.S4a-i).
Fam177a1 knockout preserved VSMCs contractile phenotype in vitro. Fam177a1WT and Fam177a1-/- rat VSMCs were stimulated with vehicle or PDGF-BB (20ng/ml). Cell proliferation was examined by CCK8 assays (a), EdU staining (b) and cell cycle flow cytometry (c) (b, scale bar: 100μm; d, scale bar: 100μm; e, scale bar: 200μm). Cell migration was examined using Transwell (d) and wound healing assays (e). f. Representative Western blotting and quantification of FAM177A1, SM22α, αSMA, CNN1, PCNA, MMP9 expression in Fam177a1WT and Fam177a1-/- rat VSMCs after PDGF-BB (20ng/ml, 48h) induction. All Data are presented as means ± SEM. P-values are calculated using the one-way ANOVA with a post hoc test of Tukey's analysis in a-f. *p <0.05, **p<0.01, ***p<0.001. CNN1, calponin-1; PCNA, proliferating cell nuclear antigen; MMP9, matrix metalloproteinase-9.
Collectively, these results establish FAM177A1 as a novel modulator of VSMC phenotypic plasticity, governing the contractile-synthetic transition. Fam177a1 knockout blocked PDGF-BB-mediated transdifferentiation by sustaining contractile programs while suppressing proliferation/migration, whereas overexpression accelerated switching toward a synthetic state.
FAM177A1 regulates VSMC mitochondrial oxidative phosphorylation and function
After establishing FAM177A1's role in orchestrating VSMC phenotypic switching both in vitro and in vivo, the underlying cellular mechanisms remained unknown. To characterize its function in VSMCs, we performed bulk mRNA sequencing on Fam177a1WT and Fam177a1-/- rat primary VSMCs. This analysis identified 1007 upregulated genes and 870 downregulated genes in Fam177a1-/- VSMCs (Fig. 5a). Gene Ontology (GO) and Gene Set Enrichment Analysis (GSEA) of differentially expressed genes revealed significant enrichment in mitochondrial pathways, particularly oxidative phosphorylation (Fig. 5b, c). Suppressed oxidative phosphorylation and enhanced glycolysis in VSMCs, is a recognized driver of phenotypic switching[45, 46]. We therefore hypothesized that FAM177A1 modulates phenotypic switching through regulation of VSMC mitochondrial function. Quantitative PCR assays confirmed expression changes in DEGs involved in oxidative phosphorylation and mitochondrial respiratory chain complexes, including GSTA2, Ndufa4, Ndufs6, Cox6b2, Cox4i2, ATP5me, and Uqcr10 in Fam177a1-/- VSMCs (Fig. 5d). To functionally assess mitochondrial respiration, we measured oxygen consumption rate (OCR) using Seahorse XF24 flux analysis. Sequential administration of inhibitors showed enhanced basal and maximal mitochondrial respiration in Fam177a1-/- VSMCs (Fig. 5e). The ATP synthase inhibitor oligomycin induced a more pronounced OCR reduction in knockout cells, indicating greater ATP-linked respiration (Fig. 5e). Furthermore, Fam177a1-/- VSMCs exhibited decreased extracellular acidification rate (ECAR) (Fig. 5f), reflecting suppressed glycolytic activity. ATP quantification assays confirmed significantly higher ATP production in Fam177a1-/- compared to Fam177a1WT controls (Fig. 5g). These collective findings demonstrate that FAM177A1 deficiency enhances mitochondrial oxidative phosphorylation while reducing glycolytic flux, thereby promoting a contractile VSMC phenotype through improved bioenergetic efficiency.
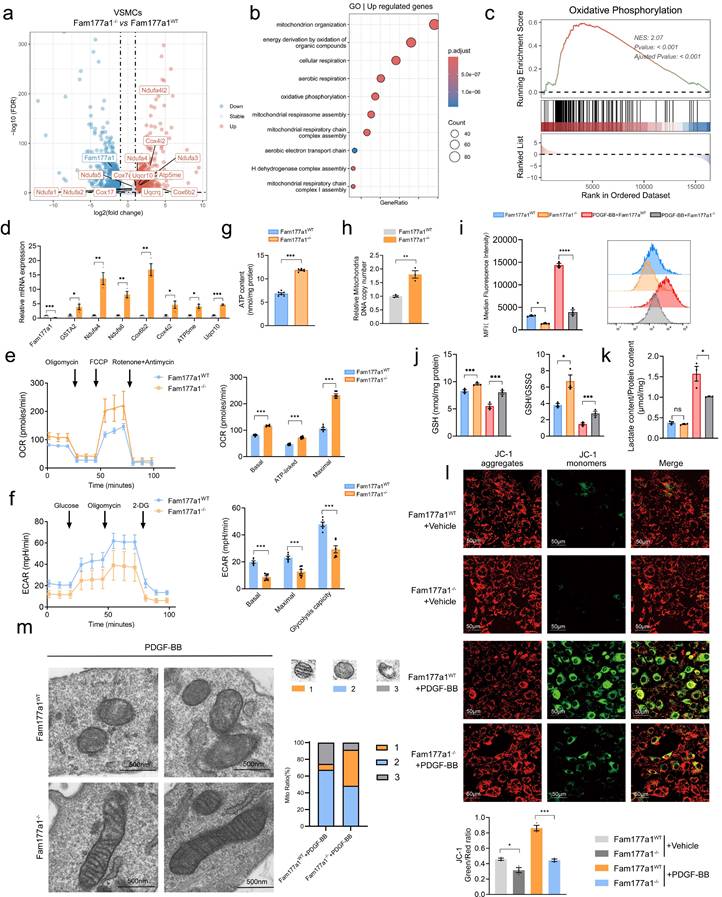
Fam177a1 knockout exhibited mitochondrial oxidative phosphorylation protective function of VSMCs. a. Volcano plot displaying differentially expressed genes (DEGs) of interest in Fam177a1WT and Fam177a1-/- rat VSMCs through transcriptome sequencing (n = 3). b. Mitochondria function related GO enrichment analysis of up-regulated genes in Fam177a1-/- VSMCs. c-d. GSEA enrichment analysis of RNA-seq data (c) and qPCR validation of mitochondria and oxidative phosphorylation related DEGs (d) (n = 3). 18S was used for control. Data were presented as relative fold change to Fam177a1WT VSMCs. e. Mitochondria oxygen consumption rate (OCR) of Fam177a1WT and Fam177a1-/- VSMCs measured using Seahorse XF24 flux analyzer. Mitochondrial effectors were added at the indicated time points (arrows, n = 3). f. Extracellular acidification rate (ECAR) of Fam177a1WT and Fam177a1-/- VSMCs measured using Seahorse XF24 flux analyzer, with sequential injections at time points (arrows, n = 3). g. Intracellular ATP production content in Fam177a1WT and Fam177a1-/- VSMCs (n = 3). h. qPCR of mtDNA copy number in VSMCs as indicated (n = 3). i. Flow cytometry analysis of ROS production in Fam177a1WT and Fam177a1-/- VSMCs after PDGF-BB (20ng/ml, 48h) induction (n = 3). j. GSH content and GSH/GSSG ratio in Fam177a1WT and Fam177a1-/- VSMCs after PDGF-BB (20ng/ml, 48h) induction (n = 3). k. Lactate content/ Protein ratio in Fam177a1WT and Fam177a1-/- VSMCs after PDGF-BB (20ng/ml, 48h) induction (n = 3). l. Representative immunofluorescence images and quantification of JC-1 staining showing membrane potential by confocal fluorescence microscopy of Fam177a1WT and Fam177a1-/- VSMCs after PDGF-BB (20ng/ml, 48h) induction (n = 3) (scale bar: 50 μm). m. Representative electron micrographs of mitochondria in Fam177a1WT and Fam177a1-/- VSMCs following PDGF-BB (20ng/ml, 48h) stimulation (scale bar: 500nm). Mitochondria were classified into three functional categories based on morphological vacuolization and cristae abundance. The proportional distribution of each mitochondrial morphology was shown in the bar graph. All Data are presented as means ± SEM. P-values are calculated using the Student's t-test (unpaired) in d-g and l and one-way ANOVA with a post hoc test of Tukey's analysis in data h-k. *p <0.05, **p<0.01, ***p<0.001. GSTA2, glutathione S-transferase alpha 2; Ndufa4, NADH dehydrogenase (ubiquinone) 1 alpha subcomplex subunit 4; Ndufs6, NADH dehydrogenase (ubiquinone) iron-sulfur protein 6; Cox6b2, Cytochrome c oxidase subunit 6B2; Cox4i2, Cytochrome c oxidase subunit 4 isoform 2; ATP5me, ATP synthase membrane subunit e; Uqcr10, Ubiquinol-cytochrome c reductase complex subunit 10; OCR, oxygen consumption rate; ECAR, extracellular acidification rate.
FAM177A1 deficiency mitigates PDGF-BB-induced mitochondrial dysfunction
To further elucidate the underlying mechanism of FAM177A1 regulating PDGF-BB-induced mitochondrial dysfunction, we performed in vitro assays to measure ROS levels, glutathione (GSH/GSSG) ratio, mitochondrial DNA (mtDNA) copy number, mitochondrial membrane potential and lactate production upon PDGF-BB stimulation. Under pathological conditions such as vascular injury, atherosclerosis, calcification, and senescence—all closely linked to mitochondrial dysfunction—vascular disease-related mitochondrial impairment during VSMC phenotypic switching accelerates disease progression[47, 48]. This impairment is characterized by suppressed oxidative phosphorylation, enhanced glycolysis, and accumulated reactive oxygen species (ROS)[49, 50]. Such mitochondrial dysfunction critically underscores the importance of mitochondrial homeostasis for restoring VSMC contractile function.
As anticipated, flow cytometry detected markedly increased ROS levels during PDGF-BB-induced phenotypic switching (Fig. 5i), indicating mitochondrial oxidative stress. Importantly, Fam177a1 knockout effectively attenuated PDGF-BB-stimulated ROS production (Fig. 5i). Assessment of cellular redox status via GSH/GSSG measurements revealed elevated GSH content and increased GSH/GSSG ratios in Fam177a1-/- VSMCs (Fig. 5j), demonstrating maintained cellular reduction capacity and protection against PDGF-BB-induced oxidative stress. Lactate content results showed that while lactate levels were slightly lower in the Fam177a1-/- VSMCs group compared to the Fam177a1WT group, this difference was not statistically significant. However, upon PDGF-BB stimulation, lactate production was significantly suppressed in the Fam177a1-/- VSMCs group. This observation aligns with the finding that Fam177a1-/- VSMCs inhibits glycolytic flux and provides further evidence that Fam177a1-/- VSMCs can suppress the glycolytic pathway, stabilize mitochondrial function, and reduce the generation of metabolic byproducts from glycolysis (Fig. 5k). Impaired oxidative phosphorylation leads to mitochondrial damage, including dissipated mitochondrial membrane potential (ΔΨm), reduced mtDNA copy number, and morphological abnormalities. Using JC-1 staining, we observed that PDGF-BB stimulation significantly dissipated ΔΨm (reduced red/green fluorescence ratio). This effect was reversed in Fam177a1-/- VSMCs, which maintained preserved ΔΨm (Fig. 5l). Additionally, Fam177a1-/- VSMCs exhibited significantly increased mtDNA content (Fig. 5h). Electron microscopy analysis also revealed that Fam177a1-/- VSMCs exhibited a higher proportion of mitochondria with intact and well-defined cristae, whereas the percentage of mitochondria containing obscured/disintegrated cristae or vacuolar structures was significantly reduced (Fig. 5m).
Collectively, these findings demonstrate that FAM177A1 deficiency enhances mitochondrial oxidative phosphorylation, upregulates mitochondrial functional genes, reduces ROS generation, maintains redox homeostasis, preserves ΔΨm, increases mtDNA biogenesis, and mitigates mitochondrial dysfunction.
FAM177A1 interacts with SOD2
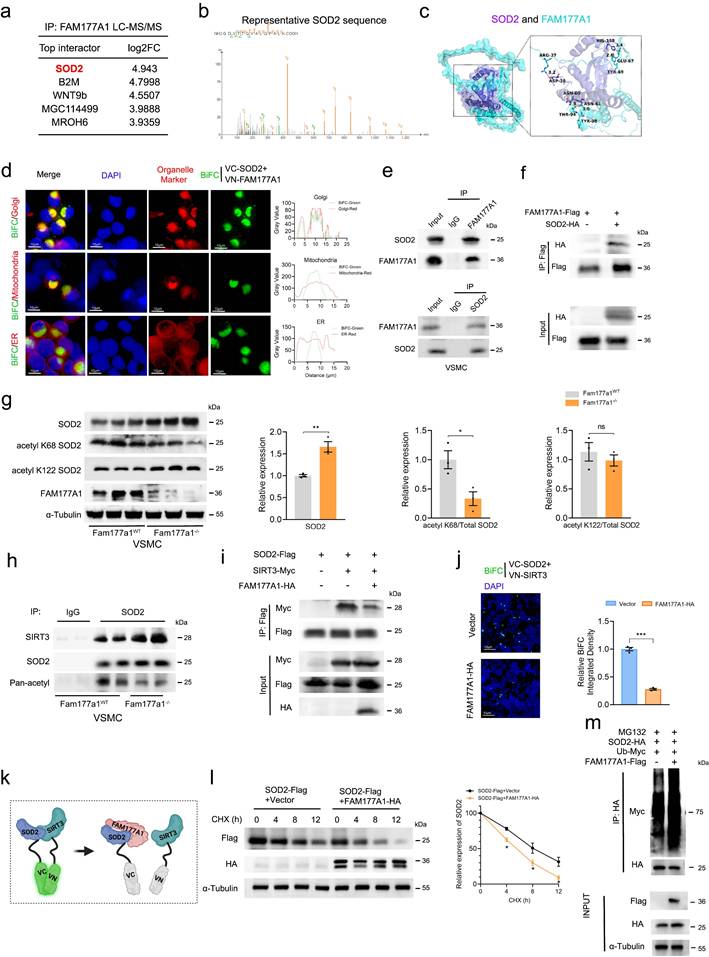
To determine how FAM177A1 regulates mitochondrial function, we performed immunoprecipitation mass spectrometry (IP-MS/MS) in VSMCs to identify its binding partners. Intriguingly, SOD2, a key mitochondrial antioxidant enzyme, emerged as the top interactor (Fig. 6a-b). Protein-protein docking of AlphaFold-predicted structures (SOD2: slate; FAM177A1: cyan) identified multiple hydrogen-bond-forming residue pairs, including FAM177A1-Ser435 and SOD2-Ser387, with a strong binding score of -640 (Fig. 6c). As FAM177A1 was poorly characterized, we examined its subcellular localization in VSMCs. Immunofluorescence revealed tight perinuclear aggregates asymmetrically positioned near the nucleus, consistent with its reported localization as a membrane-associated protein predominantly in the Golgi complex[27]. FAM177A1 co-localized perfectly with the Golgi apparatus and partially with mitochondria, but was absent from the cytoskeleton and endoplasmic reticulum (Fig. S5a). Bimolecular fluorescence complementation (BiFC) in HEK293 cells expressing SOD2-VC155 and FAM177A1-VN173 confirmed cytoplasmic interaction (Fig. S5b). This signal co-localized with Golgi and mitochondria (Fig. 6d), consistent with known SOD2/FAM177A1 distribution. Endogenous and exogenous co-immunoprecipitation validated their interaction in VSMCs and HEK293T cells, respectively (Fig. 6e-f). Truncation mapping revealed FAM177A1 binds SOD2100-180, while SOD2 binds FAM177A164-236 (Fig. S5c), aligning with AlphaFold docking predictions.
FAM177A1 interacted with SOD2 by competing with SIRT3. a-b. Top interactors of FAM177A1 identified by Liquid chromatography-tandem mass spectrometry (LC-MS/MS) (a) and the peptide fragments of SOD2 (b). c. Schematic illustration of molecular docking to predict the binding of SOD2 (purple) and FAM177A1 (blue). d. Representative immunofluorescence images of BiFC (green) and organelle marker(red) (58K for Golgi, Mitotracker for mitochondria, Calnexin for ER). Right, the co-localization analysis chart. e. The interaction of FAM177A1 and SOD2 were determined by co-IP assays in VSMCs. f. HEK293T cells were transfected with plasmids expressing SOD2 and FAM177A1 and subjected to co-IP assay. g. Representative Western blotting and quantification of SOD2, acetyl K68 SOD2, acetyl K122, FAM177A1 protein levels in Fam177a1WT and Fam177a1-/- VSMCs (n = 3). α-Tubulin was used for control. Relative expression level of SOD2, acetyl K68/total SOD2, acetyl K122/total SOD2 were shown on the analysis chart (Right). h. The interactions of SOD2 and SIRT3 were determined by co-IP assays in Fam177a1WT and Fam177a1-/- VSMCs. i. Representative Western blotting of the interactions of SOD2 and SIRT3 in HEK293T cells. j. Representative immunofluorescence images of BiFC (green) assays validating effects of FAM177A1 overexpression on the interactions of SOD2 and SIRT3 (scale bar: 10μm). k. Schematic illustration of FAM177A1 interacted with SOD2 by competing with SIRT3. l. Representative Western blotting and quantification of SOD2 in HEK293T cells transfected with plasmids expressing SOD2 and FAM177A1 followed by CHX (25μM) treatment at different time points (n = 3). m. Representative Western blotting of SOD2 polyubiquitination assays with FAM177A1overexpression in HEK293T cells. All Data are presented as means ± SEM. P-values are calculated using the Student's t-test (unpaired) analysis in g, j and l. *p <0.05, **p<0.01, ***p<0.001. LC-MS/MS, liquid chromatography-tandem mass spectrometry; BiFC, bimolecular fluorescence complementation.
FAM177A1 disrupts SIRT3-SOD2 binding to exacerbate PDGF-BB induced SOD2 suppression
In light of the above mechanistic findings, we hypothesized that FAM177A1 would lead to mitochondrial oxidative stress and impaired function by disrupting SIRT3-SOD2 binding. SOD2 scavenges superoxide produced by respiratory chain enzymes and counters mitochondrial ROS (mtROS), playing a critical role in protecting against vascular remodeling-associated mitochondrial dysfunction, including vascular stiffness, calcification, and atherosclerosis[26]. Notably, SOD2 mRNA and protein levels initially increased upon early PDGF-BB stimulation but subsequently declined, suggesting a cellular self-protective response to oxidative stress. With prolonged stimulation, SOD2 peaked at 12h and decreased progressively by 24h (Fig. S6a-b). Fam177a1-/- VSMCs resisted PDGF-BB-induced SOD2 suppression, exhibiting higher SOD2 protein levels with reduced K68 acetylation versus Fam177a1WT controls (Fig. 6g).
Since SIRT3, the primary deacetylase targeting SOD2, preserves VSMCs from oxidative stress by maintaining SOD2 activity[26, 50], we investigated whether FAM177A1 regulates SOD2 acetylation via SIRT3. However, neither overexpression nor knockout of Fam177a1 altered SIRT3 expression (Fig. S6c-d). We therefore hypothesized that FAM177A1 disrupts SIRT3-SOD2 binding. Endogenous co-IP in PDGF-BB-stimulated VSMCs (48h) confirmed reduced SIRT3-SOD2 interaction (Fig. S6e). Consistently, Fam177a1-/- VSMCs showed enhanced SIRT3-SOD2 binding and decreased acetylation level of SOD2 versus Fam177a1WT (Fig. 6h). Exogenous co-IP and BiFC in HEK293 cells further demonstrated that FAM177A1 overexpression diminished SIRT3-SOD2 interaction (Fig. 6i-k). In addition to SOD2, SIRT3 has other substrates, such as IDH2 and LCAD. To investigate whether the interaction and acetylation between SIRT3 and SOD2 are specific, we examined the acetylation levels of two other substrates, IDH2 and LCAD, in both Fam177a1WTand Fam177a1-/- VSMCs. The Western blot results showed that, unlike the findings for SOD2, the acetylation levels of IDH2 and LCAD remained unchanged in Fam177a1WTand Fam177a1-/- VSMCs, and no significant differences were observed in the protein expression levels of IDH2 and LCAD in the input lanes. This may be related to the specific binding of FAM177A1 to SOD2, which inhibits the interaction between SIRT3 and SOD2 (Fig. S6f-g). FAM177A1 overexpression also decreased SOD2 stability and promoted ubiquitin-mediated degradation (Fig. 6l-m). Using a deacetylation-mimic SOD2 K68R mutant, we found that blocking K68 acetylation reversed FAM177A1-induced SOD2 reduction and ubiquitination (Fig. S6h-i). Collectively, the binding of FAM177A1 with SOD2 impedes the SIRT3-SOD2 interaction, promoting SOD2 K68 hyperacetylation, which subsequently compromises SOD2 protein stability and leads to its degradation.
Suppression of SOD2 reversed protective effect of Fam177a1-/- on VSMCs phenotypic switch in balloon injury-induced neointimal hyperplasia or PDGF-BB stimulation models
Given FAM177A1's role as a critical regulator of the SIRT3-SOD2 axis, we inhibited SOD2 expression and activity to determine whether it functions as a primary downstream effector. Ballon-catheter injury was performed in Fam177a1WT and Fam177a1-/- rats. After ballon injury, Fam177a1-/- rats were randomly grouped and the 3-TYP group rats were injected intraperitoneally of 3-TYP (50mg/kg/d), a selective SIRT3 inhibitor once a day to inhibit SIRT3-SOD2 activity. Twenty-eight days post-injury, histological analysis (H&E and Masson staining) revealed that 3-TYP significantly reversed the protective effect of Fam177a1-/- against vascular intimal hyperplasia (Fig. 7a). The 3-TYP group showed increased neointimal area and I/M ratio with reduced lumen area versus vehicle in Fam177a1-/- rats (Fig. 7a).
SOD2 interference abrogated the effects of Fam177a1 deletion. a. Representative cross sections of H&E and Masson in male Fam177a1WT and Fam177a1-/- rats carotid arteries post ballon injury 28 days intraperitoneally injected of 3-TYP (50mg/kg/d) (scale bar:200μm). Right, quantitative analysis of the neointima/media ratio(I/M), neointima areas, media areas and lumen areas (n = 6). b. The effects of Sod2 knockdown and inhibition on cell proliferation (EdU assays, scale bar: 50μm) and cell migration (wound healing assays, scale bar: 200μm and Transwell assays, scale bar: 100μm) in Fam177a1-/- VSMCs. The EdU+/total cells ratio, wound healing rate and cell numbers per field were analyzed in the statistical chart (n = 3). c. Representative Western blotting and quantification of FAM177A1, SM22α, αSMA, SOD2 expression in Fam177a1WT and Fam177a1-/- VSMCs treated with Ad-shRNA-Scr or Ad-shRNA-SOD2. α-Tubulin was used for control. Data were presented as relative fold change to Ad-shRNA-Scr+Fam177a1WT group. d. Representative Western blotting and quantification of FAM177A1, SM22α, αSMA, acetyl K68 SOD2 expression in Fam177a1WT and Fam177a1-/- VSMCs treated with DMSO or 3-TYP (30μm). α-Tubulin was used for control. Data were presented as relative fold change toVehicle+Fam177a1WT group. All Data are presented as means ± SEM. P-values are calculated using the one-way ANOVA with a post hoc test of Tukey's analysis in a-d. *p <0.05, **p<0.01, ***p<0.001. 3-TYP, 3-(1H-1,2,3-triazol-4-yl) pyridine, selective SIRT3 inhibitor.
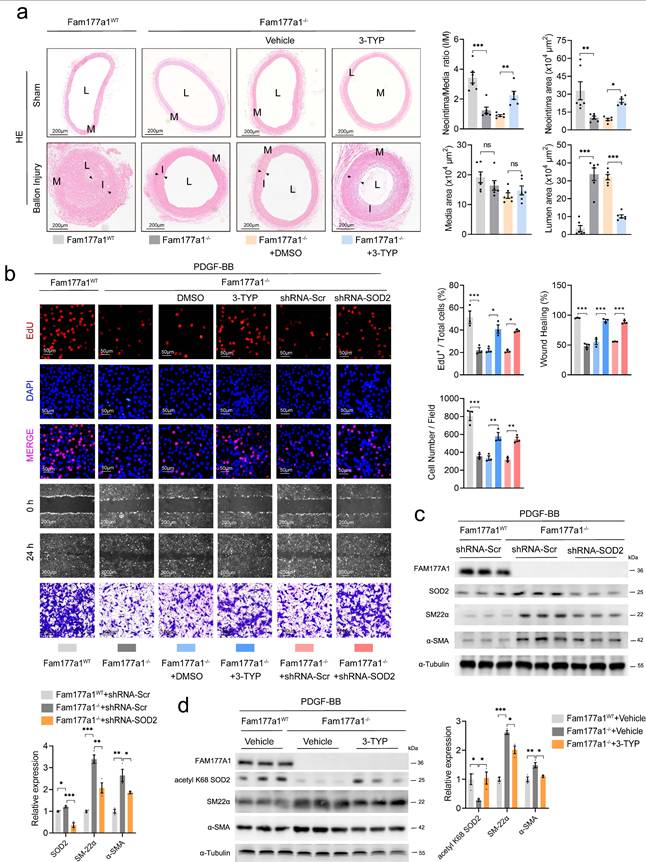
In primary VSMCs, besides pharmacological inhibition of SOD2, we knocked down Sod2 via shRNA (5′-GGAUUGAUGUGUGGGAGCACGCUUA-3′). Both approaches reversed Fam177a1-/--mediated suppression of proliferation/migration in EdU, wound healing, and Transwell assays (Fig. 7b). Western blot confirmed that 3-TYP increased SOD2 K68 acetylation, and shRNA-mediated Sod2 knockdown in Fam177a1-/- VSMCs (Fig. 7c-d). Moreover, both interventions reversed Fam177a1-/--mediated changes in contractile protein profiles (Fig. 7c-d). Measurement of ΔΨm using JC-1 probe and qPCR analysis of key mitochondrial oxidative phosphorylation genes revealed that both treatments abrogated the protective effects of Fam177a1 deficiency on mitochondrial function and oxidative phosphorylation (Fig. 8a-b).
SOD2 interference abrogated the protective effects of Fam177a1 deletion on maintain mitochondria function. a. Representative immunofluorescence images and quantification of JC-1 staining showing membrane potential by confocal fluorescence microscopy of Fam177a1WT and Fam177a1-/- VSMCs after PDGF-BB (20ng/ml, 48h) induction, with different treatment of DMSO, 3-TYP, Ad-shRNA-Scr and Ad-shRNA-SOD2 (n = 3) (scale bar: 50 μm). b. qPCR validation of mitochondria and oxidative phosphorylation related genes in Fam177a1WT and Fam177a1-/- VSMCs after PDGF-BB (20ng/ml, 48h) induction, with different treatment of DMSO, 3-TYP, Ad-shRNA-Scr and Ad-shRNA-SOD2 (n = 3). 18S was used for control. All Data are presented as means ± SEM. P-values are calculated using the one-way ANOVA with a post hoc test of Tukey's analysis in a and b. *p <0.05, **p<0.01, ***p<0.001.
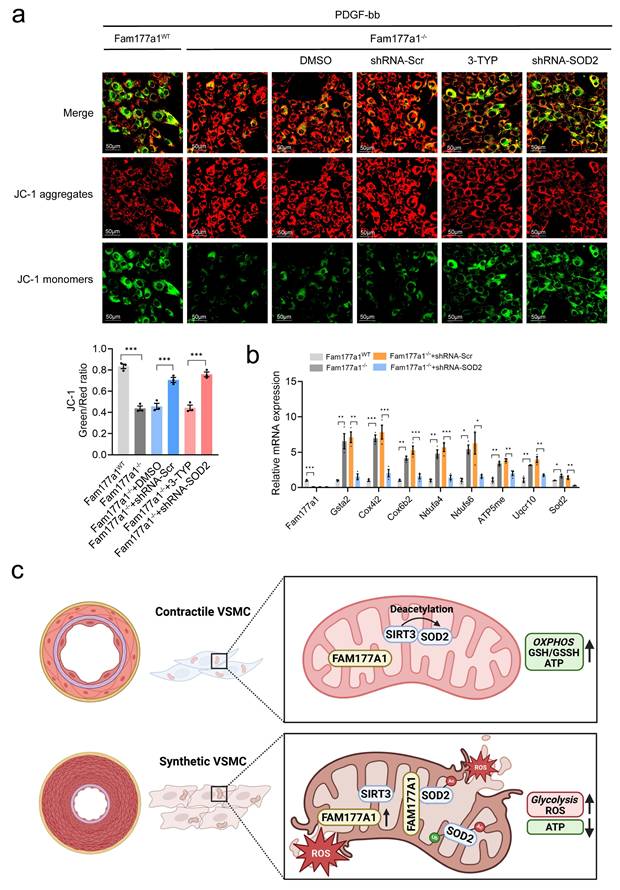
In summary, our study reveals for the first time that FAM177A1 regulates both the expression and activity of the critical antioxidant protein SOD2 in mitochondria. This mechanism modulates mitochondrial oxidative phosphorylation in VSMCs, enhancing cellular antioxidant capacity and ROS generation under vascular remodeling. Upon stimulation, increased FAM177A1 expression disrupts the SIRT3-SOD2 interaction, elevating SOD2 K68 acetylation while decreasing SOD2 activity and stability. These changes collectively impair mitochondrial function, increase mtROS production, and promote VSMC phenotypic switching and pathological vascular remodeling (Fig. 8c). Given FAM177A1's secretory nature in humans, our findings highlight its potential as a therapeutic target for vascular remodeling.
Discussion
Cardiovascular diseases remain a major global health burden, exacerbated by aging populations and metabolic disorders. VSMCs, which are essential for maintaining vascular integrity[51], undergo detrimental phenotypic switching in pathological conditions such as atherosclerosis and restenosis. This switching is characterized by abnormal proliferation, migration, and loss of contractile function[52, 53]. In recent years, there has been a growing body of research on the metabolic reprogramming of VSMCs regulating their phenotypic changes, which plays a role in various vascular diseases[54]. Mitochondria precisely regulate glucose metabolism, lipid metabolism, and amino acid metabolism through the tricarboxylic acid cycle and oxidative phosphorylation, which is crucial for maintaining metabolic homeostasis. Recent single-cell RNA sequencing studies have shown that widespread mitochondrial dysfunction occurs in various types of aortic cells and is one of the hallmarks of aortic aneurysm[55]. An increasing number of studies have also reported that genes involved in the regulation of VSMC phenotypes may influence their phenotypes through mitochondrial function[56]. Additionally, dysregulated mitochondrial homeostasis and increased glycolysis pathways lead to the involvement of glycolytic metabolites, such as lactate, in VSMC regulation. It has been reported that increased lactate can mediate elevated PARP expression and PARylation in VSMCs[48], and may also be involved in epigenetic modifications, such as participating in histone lactylation at the H3K18[57, 58] or H3K9[59] sites, thereby regulating VSMC or endothelial cell functions and contributing to processes like calcification, atherosclerosis and aortic aneurysm. While mitochondrial dysfunction in VSMCs has been implicated in these processes, the specific molecular regulators connecting mitochondrial homeostasis to phenotypic plasticity remain incompletely understood.
VSMCs are essential for maintaining vascular integrity but undergo detrimental phenotypic switching under pathological conditions, characterized by aberrant proliferation, migration, and loss of contractile function[60]. In this study, we identify FAM177A1 as a novel and critical regulator of pathological vascular remodeling through its disruption of mitochondrial redox-metabolic homeostasis. FAM177A1 is an evolutionarily conserved protein that is widely expressed in mammals and widely reported localized to the Golgi apparatus, belonging to the FAM177 protein family. Although functional studies on FAM177A1 remain limited, recent advances in molecular biology have gradually uncovered its involvement in multiple biological processes. Ko et al. elucidated a sophisticated regulatory paradigm in mouse embryonal carcinoma cells, wherein FAM177A1 expression is precisely modulated by the neural-specific microRNA miR-124a[61]. Their work established that FAM177A1 exhibited marked upregulation during proliferative phases while being potently suppressed during neuronal differentiation. Recent RNA sequencing in human fibroblast also demonstrated of FAM177A1 deficiency showed negative regulation of cell proliferation[28]. Liao et al. identified FAM177A1 as a negative regulator of IL-1β-induced signaling by inhibiting TRAF6-Ubc13 interaction[29]. Recent studies have primarily focused on the functional characterization of FAM177A1 in the nervous system. Patients with homozygous frameshift mutations in FAM177A1 typically present with early-onset neurodevelopmental abnormalities followed by neurodegenerative progression, including deteriorating motor function, refractory progressive epilepsy, arthritis/neuroinflammation, along with macrocephaly, muscular tone disorders, gait disturbance, global developmental delay, distinctive facial dysmorphism, and mild obesity[62]. In summary, although its functional roles remain incompletely characterized, emerging evidence implicates FAM177A1 in several biological processes—including cell proliferation, lipid metabolism, and innate immune response —all of which are closely associated with vascular pathological remodeling. This prompted us to investigate the role of FAM177A1 in VSMC phenotypic regulation.
We first observed significant upregulation of FAM177A1 in phenotypically modulated VSMCs within atherosclerotic lesions, following mechanical injury, or upon PDGF-BB stimulation. It has been reported that VSMCs can switch to myofibroblast-like phenotypes[63] which occurs within human AS plaque[64]. Myofibroblasts display a phenotype intermediate between fibroblasts and vascular smooth muscle cells (VSMCs), typically observed in pathological contexts. They are responsible for the production extracellular matrix components such as type I and III collagens and fibronectin—as well as secreting inflammatory cytokines[65]. Published single-cell RNA sequencing data (GSE174098) from rat carotid arteries after balloon injury demonstrated a dynamic increase in Fam177a1 expression during vascular stenosis, with particular enrichment in phenotypic switching subpopulations-Fibroblasts. Elevated FAM177A1 in Fibroblast group marked contractile VSMCs transition to a fibroblast-like state, contributing to neointima formation.
In addition, we validated the upstream transcriptional regulation of FAM177A1 using dual-luciferase reporter assays. An intriguing and unexpected finding from our promoter analysis was that, despite the well-established role of ELK1 as an upstream transcriptional activator of the c-JUN gene itself, overexpression of ELK1 did not impair the PDGF-BB-induced activation of the Fam177a1 promoter. This suggests a distinct and specific regulatory logic for the Fam177a1 gene. It appears that while the upstream PDGF-BB signal may utilize the ERK-ELK1 axis to induce c-Jun expression, the Fam177a1 promoter itself is not directly controlled by ELK1. Instead, it is selectively and directly responsive to the downstream transcription factor complex c-Jun/AP-1, which converges with the independently activated STAT3 pathway. This places FAM177A1 in a specific regulatory niche, likely as a secondary or late-response gene that integrates signals from both the AP-1 and STAT3 pathways to execute a precise transcriptional program in response to PDGF-BB, rather than being part of the immediate-early gene set directly governed by ELK1/SRF. This specificity in promoter architecture and transcription factor requirement underscores the layered complexity of the transcriptional network activated by PDGF-BB in VSMCs.
Importantly, elevated circulating levels of FAM177A1 were detected in atherosclerosis patients, highlighting its potential as a clinical biomarker. Given the presence of a secretory isoform in humans, serum FAM177A1 may serve as a non-invasive indicator of vascular remodeling progression. Consistent with this, both gain- and loss-of-function experiments, in vivo and in vitro, demonstrated that FAM177A1 drives the transition of VSMCs from a contractile to a synthetic phenotype.
Unexpectedly, immunoprecipitation mass spectrometry identified SOD2 as a major interacting partner of FAM177A1. This finding was unexpected given SOD2's well-established mitochondrial localization, which contrasts with the canonical Golgi association of FAM177A1. Subsequent validation using immunofluorescence and BiFC confirmed the dual localization of FAM177A1 to both the Golgi apparatus and mitochondria, and demonstrated strong co-localization of the FAM177A1-SOD2 complex within mitochondria. The Golgi apparatus serves as a pivotal organelle in cellular metabolism, involved in modifying, sorting, and packaging macromolecules for secretion or intracellular use[66]. It has also recently been implicated in the regulation of mitochondrial respiration, division, and dynamics[67-69]. While Golgi-derived vesicles containing PI(4)P have been recognized as important mediators of mitochondrial and ER-mitochondria crosstalk[69], protein-mediated inter-organelle interactions remain poorly understood. Thus, the subcellular distribution and regulatory mechanisms governing FAM177A1 localization clearly warrant further investigation. In addition, since the Golgi-related biological system is highly responsive to mitochondrial ROS/RNS-mediated oxidative stress and participates in damage repair[70], the current study and future investigations could significantly advance our understanding of these mechanisms.
While SIRT3-SOD2 signaling is recognized as a protective pathway against vascular oxidative stress, our data further reveal FAM177A1 acts as a unique molecular disruptor by physically interfering with SIRT3-SOD2 binding. This interference dysregulates the critical acetylation/deacetylation dynamics controlling SOD2 enzyme activity. We further observed that FAM177A1 promotes SOD2 polyubiquitination and degradation. Given FAM177A1's known interaction with the E3 ubiquitin ligase TRAF6[29], and considering other uncharacterized ligases may participate, the precise mechanisms of FAM177A1-mediated SOD2 degradation warrant further investigation. The FAM177A1-SIRT3-SOD2 axis profoundly impacts mitochondrial gene expression and function. FAM177A1 deficiency upregulated oxidative phosphorylation genes and enhanced mitochondrial respiration, while suppressing glycolytic flux. This metabolic reprogramming likely occurs through two complementary mechanisms: direct restoration of mitochondrial function, and activation of mito-nuclear communication pathways that trigger nuclear gene expression changes—creating a positive feedback loop reinforcing mitochondrial health and contractile phenotype maintenance.
Mitochondrial damage impairs the respiratory chain in VSMCs, leading to excessive ROS production and reduced ATP levels, which are also associated with VSMC proliferation, migration, and apoptosis[71]. Here, we demonstrate that pathological upregulation of FAM177A1 in vascular smooth muscle cells drives phenotypic switching by directly binding SOD2 and inhibiting its interaction with the deacetylase SIRT3. This disruption elevates SOD2 K68 acetylation, reduces SOD2 stability and activity, and ultimately causes mitochondrial dysfunction characterized by ROS accumulation, loss of membrane potential (ΔΨm), and impaired mitochondrial oxidative phosphorylation and morphology. Crucially, genetic ablation of FAM177A1 enhances SIRT3-SOD2 binding, restoring SOD2 function and promoting a contractile VSMC phenotype through coordinated improvement of redox balance and bioenergetic efficiency. Consistent protective effects across multiple models confirm that targeting FAM177A1 suppresses neointimal hyperplasia and atherosclerotic plaque progression.
Therapeutically, strategies to inhibit FAM177A1, such as monoclonal antibodies or small-molecule disruptors of FAM177A1-SOD2 binding, show significant promise. Rescue experiments using SIRT3 inhibition or SOD2 knockdown confirmed the SIRT3-SOD2 axis as the dominant downstream effector pathway. Importantly, future therapeutic development should prioritize VSMC-specific targeting to mitigate potential off-organ effects, given FAM177A1's roles in neurological and immune functions. Key unanswered questions include the differential contributions of FAM177A1 isoforms (secreted vs. membrane-bound) in vascular pathology, potential crosstalk with other SIRT3 substrates (e.g., IDH2, LCAD)[72], and identification of upstream regulators controlling FAM177A1 expression during VSMC phenotypic switching.
Conclusion
In summary, this work identifies FAM177A1 as a critical orchestrator of vascular remodeling through mitochondrial dysregulation. By disrupting SIRT3-SOD2 signaling, it initiates a self-amplifying cycle of oxidative damage and metabolic dysfunction that drives pathological VSMC phenotypic switching. Therapeutic targeting of this axis offers a dual-mechanism strategy—simultaneously improving redox balance and bioenergetic efficiency—to restore vascular homeostasis. These findings position FAM177A1 as a compelling diagnostic biomarker and therapeutic target for cardiovascular diseases characterized by vascular remodeling.
Supplementary Material
Supplementary methods, figures and tables.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No: 91949201 and No: 81830014 to Kai Huang, No: 82270489 to Cheng Wang, No: 81801062 to Xiaoguang Li). This study was funded by Health Commission Foundation of Hubei Province, China (No: WJ2023M026 to Ling Jiang). The authors express gratitude to Dr. Weifeng Zeng (Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology) for providing technical support in flow cytometry.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Xu R, Ye T, Huang W, Yue X, Morawska L, Abramson MJ. et al. Global, regional, and national mortality burden attributable to air pollution from landscape fires: a health impact assessment study. Lancet. 2024;404:2447-59
2. Timmis A, Aboyans V, Vardas P, Townsend N, Torbica A, Kavousi M. et al. European Society of Cardiology: the 2023 Atlas of Cardiovascular Disease Statistics. Eur Heart J. 2024;45:4019-62
3. Rajagopalan S, McAlister S, Jay J, Pham RD, Brook RD, Nasir K. et al. Environmental sustainability in cardiovascular practice: current challenges and future directions. Nat Rev Cardiol. 2025;22:241-54
4. Lear SA, McKee M, Hystad P, Byron Walker B, Murphy A, Brauer M. et al. Social factors, health policy, and environment: implications for cardiovascular disease across the globe. Eur Heart J. 2025
5. Pan H, Xue C, Auerbach BJ, Fan J, Bashore AC, Cui J. et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation. 2020;142:2060-75
6. Chakraborty A, Li Y, Zhang C, Li Y, Rebello KR, Li S. et al. Epigenetic Induction of Smooth Muscle Cell Phenotypic Alterations in Aortic Aneurysms and Dissections. Circulation. 2023;148:959-77
7. Yap C, Mieremet A, de Vries CJM, Micha D, de Waard V. Six Shades of Vascular Smooth Muscle Cells Illuminated by KLF4 (Krüppel-Like Factor 4). Arterioscler Thromb Vasc Biol. 2021;41:2693-707
8. Xu W, Wu Y, Mao R, Jia Y, Jiang H, Zhang F. et al. Poly(ADP-ribose) polymerase 1 orchestrates vascular smooth muscle cell homeostasis in arterial disease. Exp Mol Med. 2025
9. Song T, Zhao S, Luo S, Chen C, Liu X, Wu X. et al. SLC44A2 regulates vascular smooth muscle cell phenotypic switching and aortic aneurysm. J Clin Invest. 2024 134
10. Qi M, Xin S. FGF signaling contributes to atherosclerosis by enhancing the inflammatory response in vascular smooth muscle cells. Mol Med Rep. 2019;20:162-70
11. Karvinen H, Rutanen J, Leppänen O, Lach R, Levonen AL, Eriksson U. et al. PDGF-C and -D and their receptors PDGFR-alpha and PDGFR-beta in atherosclerotic human arteries. Eur J Clin Invest. 2009;39:320-7
12. Beg MA, Huang M, Vick L, Rao KNS, Zhang J, Chen Y. Targeting mitochondrial dynamics and redox regulation in cardiovascular diseases. Trends Pharmacol Sci. 2024;45:290-303
13. Su B, Mitra S, Gregg H, Flavahan S, Chotani MA, Clark KR. et al. Redox regulation of vascular smooth muscle cell differentiation. Circ Res. 2001;89:39-46
14. Badran A, Nasser SA, Mesmar J, El-Yazbi AF, Bitto A, Fardoun MM. et al. Reactive Oxygen Species: Modulators of Phenotypic Switch of Vascular Smooth Muscle Cells. Int J Mol Sci. 2020 21
15. Monteiro R, Azevedo I. Chronic inflammation in obesity and the metabolic syndrome. Mediators Inflamm. 2010. 2010
16. Clempus RE, Griendling KK. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc Res. 2006;71:216-25
17. Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296-9
18. Branchetti E, Poggio P, Sainger R, Shang E, Grau JB, Jackson BM. et al. Oxidative stress modulates vascular smooth muscle cell phenotype via CTGF in thoracic aortic aneurysm. Cardiovasc Res. 2013;100:316-24
19. Velarde V, de la Cerda PM, Duarte C, Arancibia F, Abbott E, González A. et al. Role of reactive oxygen species in bradykinin-induced proliferation of vascular smooth muscle cells. Biol Res. 2004;37:419-30
20. Zhang Z, Zhao X, Zhang X, Wan S, An P, Zhu Y. et al. Mitochondrial cardiovascular diseases: molecular mechanisms, multi-omics exploration and therapeutic strategies. J Adv Res. 2025
21. Li J, Li X, Song S, Sun Z, Li Y, Yang L. et al. Mitochondria spatially and temporally modulate VSMC phenotypes via interacting with cytoskeleton in cardiovascular diseases. Redox Biol. 2023;64:102778
22. Jia Y, Mao C, Ma Z, Huang J, Li W, Ma X. et al. PHB2 Maintains the Contractile Phenotype of VSMCs by Counteracting PKM2 Splicing. Circ Res. 2022;131:807-24
23. Roldán-Montero R, Pérez-Sáez JM, Cerro-Pardo I, Oller J, Martinez-Lopez D, Nuñez E. et al. Galectin-1 prevents pathological vascular remodeling in atherosclerosis and abdominal aortic aneurysm. Sci Adv. 2022;8:eabm7322
24. Paredes F, Williams HC, Liu X, Holden C, Bogan B, Wang Y. et al. The mitochondrial protease ClpP is a druggable target that controls VSMC phenotype by a SIRT1-dependent mechanism. Redox Biol. 2024;73:103203
25. Miao G, Zhao X, Chan SL, Zhang L, Li Y, Zhang Y. et al. Vascular smooth muscle cell c-Fos is critical for foam cell formation and atherosclerosis. Metabolism. 2022;132:155213
26. Dikalova AE, Itani HA, Nazarewicz RR, McMaster WG, Flynn CR, Uzhachenko R. et al. Sirt3 Impairment and SOD2 Hyperacetylation in Vascular Oxidative Stress and Hypertension. Circ Res. 2017;121:564-74
27. Ugur B, Schueder F, Shin J, Hanna MG, Wu Y, Leonzino M. et al. VPS13B is localized at the interface between Golgi cisternae and is a functional partner of FAM177A1. J Cell Biol. 2024 223
28. Kohler JN, Legro NR, Baldridge D, Shin J, Bowman A, Ugur B. et al. Loss of function of FAM177A1, a Golgi complex localized protein, causes a novel neurodevelopmental disorder. Genet Med. 2024;26:101166
29. Liao BW, Zhang HY, Du WT, Ran Y, Wang YY, Xu ZS. FAM177A1 Inhibits IL-1β-Induced Signaling by Impairing TRAF6-Ubc13 Association. J Immunol. 2021;207:3090-7
30. Chen H, Guo M, Yue D, Zhao J, Zhou Y, Chen C. et al. MicroRNA-7 negatively regulates Toll-like receptor 4 signaling pathway through FAM177A. Immunology. 2021;162:44-57
31. Yang H, Chen L, Liu Y. A large-scale plasma proteome Mendelian randomization study identifies novel causal plasma proteins related to primary biliary cholangitis. Front Immunol. 2023;14:1052616
32. Parada H Jr, Sun X, Fleming JM, Williams-DeVane CR, Kirk EL, Olsson LT. et al. Race-associated biological differences among luminal A and basal-like breast cancers in the Carolina Breast Cancer Study. Breast Cancer Res. 2017;19:131
33. Jia Y, Wang M, Mao C, Yu F, Wang Y, Xiao R. et al. COMP-prohibitin 2 interaction maintains mitochondrial homeostasis and controls smooth muscle cell identity. Cell Death Dis. 2018;9:676
34. Zhu N, Guo ZF, Kazama K, Yi B, Tongmuang N, Yao H. et al. Epigenetic regulation of vascular smooth muscle cell phenotypic switch and neointimal formation by PRMT5. Cardiovasc Res. 2023;119:2244-55
35. Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL. et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med. 2019;25:1280-9
36. Frismantiene A, Philippova M, Erne P, Resink TJ. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell Signal. 2018;52:48-64
37. Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012;95:156-64
38. Cao G, Xuan X, Hu J, Zhang R, Jin H, Dong H. How vascular smooth muscle cell phenotype switching contributes to vascular disease. Cell Commun Signal. 2022;20:180
39. Teufel F, Almagro Armenteros JJ, Johansen AR, Gíslason MH, Pihl SI, Tsirigos KD. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat Biotechnol. 2022;40:1023-5
40. Davies MJ, Richardson PD, Woolf N, Katz DR, Mann J. Risk of thrombosis in human atherosclerotic plaques: role of extracellular lipid, macrophage, and smooth muscle cell content. Br Heart J. 1993;69:377-81
41. Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM. et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628-37
42. Miano JM, Fisher EA, Majesky MW. Fate and State of Vascular Smooth Muscle Cells in Atherosclerosis. Circulation. 2021;143:2110-6
43. Bentzon JF, Majesky MW. Lineage tracking of origin and fate of smooth muscle cells in atherosclerosis. Cardiovasc Res. 2018;114:492-500
44. Shi J, Yang Y, Cheng A, Xu G, He F. Metabolism of vascular smooth muscle cells in vascular diseases. Am J Physiol Heart Circ Physiol. 2020;319:H613-h31
45. Vásquez-Trincado C, García-Carvajal I, Pennanen C, Parra V, Hill JA, Rothermel BA. et al. Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol. 2016;594:509-25
46. Pearce WJ. Mitochondrial influences on smooth muscle phenotype. Am J Physiol Cell Physiol. 2024;326:C442-c8
47. Yang L, Gao L, Nickel T, Yang J, Zhou J, Gilbertsen A. et al. Lactate Promotes Synthetic Phenotype in Vascular Smooth Muscle Cells. Circ Res. 2017;121:1251-62
48. Zhu Y, Zhang JL, Yan XJ, Ji Y, Wang FF. Exploring a new mechanism between lactate and VSMC calcification: PARP1/POLG/UCP2 signaling pathway and imbalance of mitochondrial homeostasis. Cell Death Dis. 2023;14:598
49. Cao K, Zhang T, Li Z, Song M, Li A, Yan J. et al. Glycolysis and de novo fatty acid synthesis cooperatively regulate pathological vascular smooth muscle cell phenotypic switching and neointimal hyperplasia. J Pathol. 2023;259:388-401
50. You M, Wang B, Li L, Liu M, Wang L, Cao T. et al. SIRT3 Represses Vascular Remodeling via Reducing Mitochondrial Ac-CoA Accumulation in Vascular Smooth Muscle Cells. Arterioscler Thromb Vasc Biol. 2025;45:985-1005
51. Wissler RW. The arterial medial cell, smooth muscle, or multifunctional mesenchyme? Circulation. 1967;36:1-4
52. Liu M, Gomez D. Smooth Muscle Cell Phenotypic Diversity. Arterioscler Thromb Vasc Biol. 2019;39:1715-23
53. Zhu M, Zhu W, Pan J, Chen Z, Yan Y, Wang S. et al. Fibroblast Growth Factor 21 Promotes Vascular Smooth Muscle Cell Contractile Polarization via p38 Mitogen-Activated Protein Kinase-Promoted Serum Response Factor Phosphorylation. Research (Wash D C). 2025;8:0815
54. Wu Z, Lu H, Liu T, Yue X, Wang L, Ma C. et al. ZFP36 Protects against Abdominal Aortic Aneurysm Formation by Regulating Vascular Smooth Muscle Phenotypic Switch. Research (Wash D C). 2026;9:1078
55. Li Y, Ren P, Dawson A, Vasquez HG, Ageedi W, Zhang C. et al. Single-Cell Transcriptome Analysis Reveals Dynamic Cell Populations and Differential Gene Expression Patterns in Control and Aneurysmal Human Aortic Tissue. Circulation. 2020;142:1374-88
56. Sun LY, Lyu YY, Zhang HY, Shen Z, Lin GQ, Geng N. et al. Nuclear Receptor NR1D1 Regulates Abdominal Aortic Aneurysm Development by Targeting the Mitochondrial Tricarboxylic Acid Cycle Enzyme Aconitase-2. Circulation. 2022;146:1591-609
57. Ma W, Jia K, Cheng H, Xu H, Li Z, Zhang H. et al. Orphan Nuclear Receptor NR4A3 Promotes Vascular Calcification via Histone Lactylation. Circ Res. 2024;134:1427-47
58. Li YE, Liu S, Wang L, Du Y, Wu L, Chen H. et al. March2 Alleviates Aortic Aneurysm/Dissection by Regulating PKM2 Polymerization. Circ Res. 2025;136:e73-e93
59. Ma Y, Feng S, Jiang Y, Jiang J, Liu R, Ma Y. et al. SLC22A6-dependent lactylation of H3K9 aggravates endothelial dysfunction and atherosclerosis. Metabolism. 2026;175:156426
60. Cao X, Jia J, Gao Q, Tao J, Wei M, Song Y. et al. Elevated Dickkopf 3 Promotes Abdominal Aortic Aneurysm Formation via Modulated Phenotype Switch of Vascular Smooth Muscle Cells. Research (Wash D C). 2025;8:0873
61. Ko HY, Lee DS, Kim S. Noninvasive imaging of microRNA124a-mediated repression of the chromosome 14 ORF 24 gene during neurogenesis. Febs j. 2009;276:4854-65
62. Alazami AM, Patel N, Shamseldin HE, Anazi S, Al-Dosari MS, Alzahrani F. et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015;10:148-61
63. Rønnov-Jessen L, Petersen OW, Koteliansky VE, Bissell MJ. The origin of the myofibroblasts in breast cancer. Recapitulation of tumor environment in culture unravels diversity and implicates converted fibroblasts and recruited smooth muscle cells. J Clin Invest. 1995;95:859-73
64. Hao H, Gabbiani G, Camenzind E, Bacchetta M, Virmani R, Bochaton-Piallat ML. Phenotypic modulation of intima and media smooth muscle cells in fatal cases of coronary artery lesion. Arterioscler Thromb Vasc Biol. 2006;26:326-32
65. Van Linthout S, Miteva K, Tschöpe C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc Res. 2014;102:258-69
66. Jiang Z, Hu Z, Zeng L, Lu W, Zhang H, Li T. et al. The role of the Golgi apparatus in oxidative stress: is this organelle less significant than mitochondria? Free Radic Biol Med. 2011;50:907-17
67. Jung Y, Artan M, Kim N, Yeom J, Hwang AB, Jeong DE. et al. MON-2, a Golgi protein, mediates autophagy-dependent longevity in Caenorhabditis elegans. Sci Adv. 2021;7:eabj8156
68. Zhong YL, Xu CQ, Li J, Liang ZQ, Wang MM, Ma C. et al. Mitochondrial dynamics and metabolism in macrophages for cardiovascular disease: A review. Phytomedicine. 2025;140:156620
69. Tábara LC, Morris JL, Prudent J. The Complex Dance of Organelles during Mitochondrial Division. Trends Cell Biol. 2021;31:241-53
70. Jiang Z, Hu Z, Zeng L. et al. The role of the Golgi apparatus in oxidative stress: is this organelle less significant than mitochondria? Free Radic Biol Med. 2011;50:907-17
71. Grootaert MOJ, Finigan A, Figg NL, Uryga AK, Bennett MR. SIRT6 Protects Smooth Muscle Cells From Senescence and Reduces Atherosclerosis. Circ Res. 2021;128:474-91
72. Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB. et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121-5
Author contact
![]() Corresponding authors: Email: huangkai1edu.cn, lxgedu.cn and cwangunionedu.cn.
Corresponding authors: Email: huangkai1edu.cn, lxgedu.cn and cwangunionedu.cn.