Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
E2s in the hallmarks of cancer
E2s in enabling characteristics...
Targeting E2s to treat cancer
Conclusions and Perspective
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(6):3244-3271. doi:10.7150/ijbs.130297 This issue Cite
Review
Ubiquitin-conjugating Enzymes in Cancer
Zhiyang Yao1,2, Ti Peng2,3, Hao Dong2, Yong Liao1,2, Kai Miao4 ![]() , Jiang-Jiang Qin1,2,3
, Jiang-Jiang Qin1,2,3 ![]() , Xiaoqing Guan5
, Xiaoqing Guan5 ![]()
1. College of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou, Zhejiang, 310014, China.
2. Center for Innovative Drug Research, Hangzhou Institute of Medicine (HIM), Chinese Academy of Sciences, Hangzhou, Zhejiang, 310018, China.
3. School of Molecular Medicine, Hangzhou Institute for Advanced Study, UCAS, Hangzhou, Zhejiang, 310024, China.
4. MOE Frontier Science Centre for Precision Oncology, University of Macau, Macau SAR, 999078, China.
5. Department of Hepato-Pancreato-Biliary & Gastric Medical Oncology, Zhejiang Cancer Hospital, Hangzhou, Zhejiang, 310022, China.
Received 2025-12-20; Accepted 2026-2-12; Published 2026-3-4
Abstract

Ubiquitin-conjugating enzymes (E2s) are emerging as critical regulators of oncogenic signaling networks by modulating protein stability, localization, and interactome dynamics. Distinct E2s orchestrate ubiquitin chain topology to drive cancer hallmarks including proliferation, immune evasion, metastasis, and metabolic reprogramming. Despite their central role in the ubiquitin-proteasome system, E2s remain underexploited as therapeutic targets. This review systematically maps E2-mediated signaling mechanisms across cancer types and proposes translational strategies for E2-targeted intervention.
Keywords: E2 ubiquitin-conjugating enzyme, cancer, target, mechanism.
Introduction
Cancer remains a leading cause of global mortality, with >20 million new cases projected in 2025. Despite the success of kinase and immune-checkpoint inhibitors, most tumors eventually evade these agents through feedback reactivation of downstream signaling circuits [1-4], highlighting the urgent need to target alternative signaling layers that orchestrate oncogenic fate decisions. Targeting ubiquitination has emerged as an evolvable and druggable signaling stratum that writes, erases and re-wires oncogenic pulses.
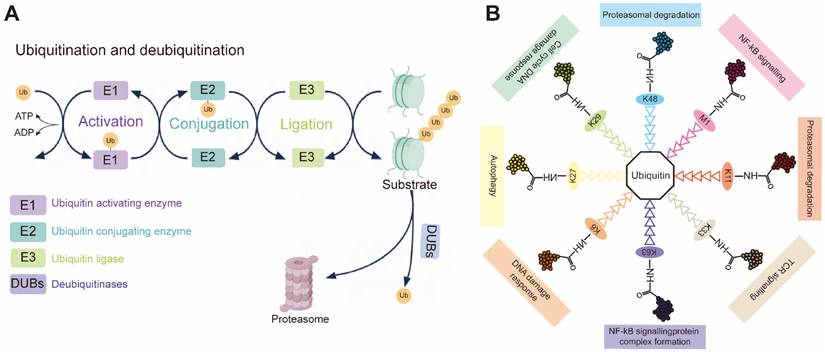
During ubiquitination, a cascade of E1 (activating), E2 (conjugating) and E3 (ligase) enzymes sequentially installs ubiquitin on substrates (Figure 1A). First, an E1-ubiquitin thioester bond is formed between the C-terminal Gly carboxyl group of ubiquitin and the active site Cys of the E1 through an ATP-dependent reaction. E1 then transfers the activated ubiquitin to the Cys residue of the E2 to form an E2-ubiquitin thioester linker intermediate by transesterification. Eventually, the E2-ubiquitin complex associates with an E3 ligase to deliver ubiquitin to a lysine, serine, threonine or even non-protein substrate [5, 6]. In reality, the E2 step is a biochemical switchboard: each of the ~40 human members [7] selects which of eight possible chain linkages (K6, K11, K27, K29, K33, K48, K63 or M1) will be forged and how long or branched the resulting polymer will be (Figure 1B) [8, 9]. Previous studies have shown that K11 and K48 chains lead to the degradation by the 26S proteasome, and that K63 chain is involved in various intracellular non-proteolytic functions, such as autophagy, DNA repair, and cell cycle control [10].
(A) Schematic demonstration of the ubiquitination process. Upon ATP-dependent activation, ubiquitin (Ub) is transferred from the E1 activating enzyme to the E2 conjugating enzyme. The E3 ubiquitin ligase then facilitates the final transfer and covalent attachment of ubiquitin to a specific substrate protein. The polyubiquitinated target is ultimately recognized and degraded by the 26S proteasome complex. (B) Individual ubiquitin linkage types. Ubiquitin a small 76-amino acid protein, can be attached to a targeted substrate or a ubiquitin molecule that is already attached to a substrate, resulting in specific polyubiquitin linkage types.
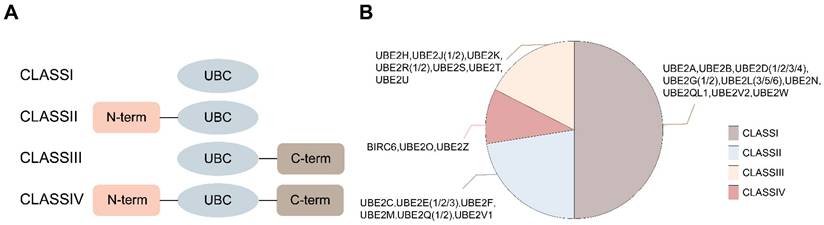
E2s are structurally categorized into four classes: Class I (UBC catalytic core only), Class II (with an N-terminal extension), Class III (with a C-terminal extension), and Class IV (with both N- and C-terminal extensions) (Figure 2) [11]. Most E2s can interact with E1 and one or more E3s, thereby providing a binding platform for E1, E3, and activated ubiquitin/ubiquitin-like (Ubl) proteins [12]. Additionally, some E2s participate in non-canonical ubiquitin transfer processes, such as regulating the activity of other enzymes [13]. Although all E2s share a conserved catalytic core composed of four antiparallel β-strands flanked by four α-helices, this structure can mediate diverse biological functions, including substrate degradation, scaffolding complex assembly, and even the formation of degradation-resistant ubiquitin chains that regulate kinase clustering, DNA repair foci assembly, or NF-κB signalosome activation.
Classification of E2s. (A) Four classes of E2s are depicted in different framed boxes upon the absence (class I) or presence of additional extensions in the N- or C-terminal of the UBC domain (class II or class III, respectively) or in both termini (class IV). (B) All specific classifications of E2s.
The functional diversity of E2s is largely determined by their extension regions beyond the conserved catalytic core domain. These extensions serve as key structural determinants influencing E2 subcellular localization, the stability of their interactions with E1, and the regulation of E3 ligase activity. Taking the N-terminal extension as an example, it enables fine-tuning of E2 interactions with both E1 and E3. The N-terminal region of UBE2M enhances its binding to the NEDD8-specific E1 while reducing its affinity for the ubiquitin-specific E1 [14]. Similarly, the N-terminal extensions of E2s like UBE2C also modulate ubiquitin transfer efficiency by decreasing their affinity for E1 [15]. This structural feature may act cooperatively with the E2-E3 interaction interface to facilitate the ubiquitination of substrates lacking canonical degradation signals. Research indicates that deletion of the N-terminus in UBE2C leads to excessive ubiquitin transfer mediated by its E3 ligase APC/C, resulting in aberrant substrate degradation [15]. Furthermore, the N-terminal region directly participates in E3 ligase recognition. For instance, UBE2L3 utilizes Arg5 and Arg15 within its N-terminal α1-helix to bind the E3 ligase CBL, thereby negatively regulating receptor tyrosine kinase signaling [16]. The interaction between UBE2D2 and the E3 ligase CNOT4 depends on Lys4 within its N-terminal α1-helix [17].
On the other hand, C-terminal extensions are primarily involved in regulating enzymatic activity and ubiquitin chain elongation. For example, the C-terminal extension of UBE2R1 is essential for polyubiquitin chain formation, with recent studies suggesting its mechanism may involve interactions between C-terminal residues and the conjugated ubiquitin molecule [19]. The acidic C-terminal tail of UBE2R2 can be phosphorylated by protein kinase CK2. This modification can affect SCF complex-mediated Sic1 ubiquitination, cell cycle progression, and potentially the intracellular distribution of this E2 [20]. Extension regions also play a crucial role in subcellular localization. Although UBE2G2 itself lacks a transmembrane domain, its C-terminal extension can specifically recognize the endoplasmic reticulum (ER) membrane protein Cue1. This interaction "anchors" UBE2G2 to the ER membrane, enabling efficient collaboration with ER-associated E3 ligases [21]. Similarly, phosphorylation of the C-terminal tail of UBE2R2 is also involved in regulating its intracellular localization [22].
E2s are not only key pathogenic factors in neurodegenerative diseases [11], chromosomal instability syndromes [23], and autoimmune disorders [24], but also are implicated in every cancer hallmark, promoting cancer initiation and progression. E2 dysregulation has been documented in a wide spectrum of cancers. Moreover, numerous E2s drive cell cycle progression, DNA repair, and stimulate oncogenic signaling pathways during malignant transformation. Consequently, E2s are increasingly recognized as valuable cancer-related biomarkers for diagnosis, prognosis, and therapeutic targeting [25].
Here, we synthesize current knowledge on how E2s sculpt the linkage-specific transduction networks that drive each hallmark of cancer. We then inventory existing small-molecule inhibitors and miRNA mimics/antagomirs that directly engage E2s or their 3'-UTRs, summarizing their selectivity and in vitro and in vivo efficacy. Finally, we chart emerging research directions that will shape the next decade of E2-centric clinical development.
E2 mutations in cancer
Gene mutations [26, 27] are one of the fundamental drivers of biological phenotypic diversity and disease onset. Mutations are broadly categorized into germline mutations and somatic mutations [28]: Germline mutations are heritable and constitute the root cause of numerous genetic disorders; whereas somatic mutations, which occur in somatic cells, are a key driver in tumorigenesis and progression. In cancer research, analyzing mutation profiles in specific genes—such as proto-oncogenes and tumor suppressor genes—not only reveals the molecular mechanisms of tumorigenesis but also provides critical theoretical foundations for targeted therapies and biomarker development. The subject of this paper, E2s exhibit relatively rare mutations. However, these mutations play pivotal roles in the onset and progression of various cancers. Different types of E2 mutations disrupt normal catalytic function, substrate recognition, or protein interactions, leading to specific pathological processes.
UBE2A: Somatic mutations in the UBE2A gene at two nonadjacent residues (D114V and I33M) have been identified in CML. These mutations cause conformational changes in the UBE2A protein, resulting in the loss of its ability to regulate the activity of downstream target proteins. Compared to the WT, the mutant UBE2A exhibits significantly reduced ATP consumption and correspondingly weaker ubiquitin-binding activity, leading to a loss of regulatory control over CSF3R transcription. CSF3R, a key regulator of myeloid lineage differentiation and development [29], is affected by UBE2A dysfunction, which may hinder normal cell differentiation by interfering with granulocyte development-related pathways [30]. Notably, UBE2A mutations are not exclusive to CML. They have also been observed in patients with DLBCL [31]. In Chinese DLBCL cases, UBE2A mutations account for approximately 10% of instances and are found exclusively in patients with bone marrow involvement [32]. Furthermore, these mutations are significantly associated with a shortened time to transformation into an aggressive FL subtype following biopsy confirmation [33]. Additionally, UBE2A has been reported as a potential novel driver gene in cervical cancer [34]. This cross-cancer evidence suggests that interventions targeting UBE2A may represent a potential auxiliary therapeutic strategy, although the specific mechanisms underlying its oncogenic role require further elucidation.
UBE2C: UBE2C is widely overexpressed in multiple cancer cell lines and tissues, and its elevated expression is significantly associated with tumor proliferation, invasion, metastasis, and poor clinical prognosis [35]. Mutations in this gene are commonly observed in various solid tumors, including CRC, EC, GC, BCa, NSCLC, SARC, MEL, PC, and GBM. Pan-cancer analysis based on the cBioPortal database reveals diverse forms of genetic alterations in UBE2C, primarily including gene amplification, point mutations, deep deletions, and multiple alterations, with gene amplification being the most predominant variant type [36]. These genetic alterations, particularly mutations and amplifications, are considered key factors driving the abnormally high expression of UBE2C in tumors such as UCEC. In PRAD, CNVs of UBE2C are significantly associated with patients' progression-free survival (PFS), disease-specific survival (DSS), and disease-free survival (DFS), indicating its important prognostic value. Furthermore, abnormal mRNA expression of UBE2C is mainly associated with missense mutations and splicing mutations, which are predominantly distributed in diploid regions and copy number gain regions of the genome [36].
UBE2G2: Menezes et al. identified deleterious mutations of UBE2G2 in human leukemia for the first time [37]. Targeted resequencing results revealed a single-amino-acid-change mutation in the UBE2G2 gene, specifically an allele T>A variation, with a mutation frequency of 4% in the study cohort. The impact of this mutation on downstream signaling pathways requires further investigation.
UBE2I: UBE2I regulates the proliferation and transformation of different cancers and is targeted by a variety of viruses, including HIV, EBV, and HPV [38, 39]. A large number of novel missense mutations in UBE2I have been identified in cancer patients, but their functional implications are unclear [40, 41].
UBE2M: SNVs or CNVs in the UBE2M gene can be observed across multiple cancer types [42]. Particularly in LUSC, SNVs in UBE2M correlate with lower patient survival rates, while its CNVs also indicate poorer clinical prognosis. Mutated UBE2M modulates neddylation pathways by regulating NEDD8 binding, thereby contributing to tumorigenesis and progression. Functionally, UBE2M primarily promotes cell cycle progression while inhibiting RAS/MAPK and RTK signaling pathways.
UBE2O: Mutations in UBE2O are relatively common across multiple cancers, including breast, gastric, renal, and ovarian cancers [43]. Analysis of data from The Cancer Genome Atlas (TCGA) database indicates that UBE2O gene amplification frequently occurs in various cancer types, such as gastric and lung cancers. This amplification drives cancer cell proliferation by promoting the ubiquitination and degradation of AMPKα2 [44]. UBE2O mutations, including potential splicing variants and other genetic defects, may impact its biological functions, such as regulation of cell proliferation and apoptosis. However, the role of UBE2O mutations in cancer initiation and progression remains to be fully explored.
UBE2S: Huijie Bian [45] et al. identified two novel somatic mutations in UBE2S (p.Gly57Ala and p.Lys63Asn) via whole-exome sequencing. Although researchers have not yet investigated the molecular mechanisms of these mutant UBE2S variants in depth, scale-invariant feature transformation analysis predicted functional effects, suggesting that these two amino acid substitutions may have adverse consequences.
UBE2T: Analysis of large-scale genomic and expression profiling data covering hundreds of cancer cell lines in the cBioPortal platform reveals that the UBE2T gene is altered in multiple cancers, with particularly prominent high-frequency amplification [46]. Taking breast cancer as an example, UBE2T exhibits significant overexpression at both the transcriptional and protein levels [47]. Its potential oncogenic mechanism may involve polyubiquitination modification of the E3 ligase RING ligase BRCA1—a key tumor suppressor gene in breast cancer development—and subsequent promotion of its degradation via the proteasome pathway. Furthermore, a novel frameshift mutation c.415_418insAGCC in UBE2T was identified in germ cells from breast/ovarian cancer patients [48]. This mutation introduces 22 additional amino acids downstream, resulting in loss-of-function protein.
UBE2W: Yan Yuan et al. [49] found that 20% of BC patients exhibited alterations in the UBE2W gene, with significant differences in mRNA expression levels across all analyzed samples. Further analysis revealed markedly elevated mRNA expression in cases with UBE2W gene amplification. Additionally, patients harboring UBE2W mutations exhibited significantly shorter overall survival and recurrence-free survival compared to those without this genetic alteration. These findings suggest that UBE2W may influence clinical outcomes in BC patients by interacting with the E3 ligase RBX1.
The pathogenic significance of E2 mutations extends far beyond their low frequency in pan-cancer genomic landscapes. While these mutations occur sparsely, they can function as potent drivers of oncogenesis through diverse mechanisms—including catalytic dysregulation, dosage-dependent effects, and altered protein interaction interfaces. Consequently, E2 mutations may serve as critical determinants of tumor progression, therapy resistance, and immune evasion in specific contexts. However, the direct role of these mutations in cancer progression has not been fully explored.
Even mutations that appear infrequently in pan-cancer genomic maps may carry significant pathogenic implications. For instance, IDH1/2 mutations occur as low-frequency missense mutations in gliomas and acute myeloid leukemia, yet their mutation sites are located at the enzyme's active site, directly impacting its catalytic properties [50]. IDH1/2 mutations reduce α-ketoglutarate production capacity and confer an abnormal function—converting α-ketoglutarate into (R)-2-hydroxyglutarate ((R)-2HG) using NADPH as a cofactor [51, 52]. (R)-2HG is considered a primary oncogenic metabolite, exerting multiple biological effects in IDH1/2-mutant tumors. Consequently, IDH1/2 mutations are regarded as gain-of-function alterations [51]. Similarly, under specific contexts, E2 mutations may serve as critical determinants of tumor progression, therapeutic resistance, and immune evasion. However, the mechanisms underlying the role of these E2 mutations in cancer progression remain poorly understood.
E2s in the hallmarks of cancer
The occurrence and development of cancer is a complex process that involves a variety of factors and mechanisms. The publication of the study [159] by Douglas Hanahan and Robert A. Weinberg et al. describing the top ten hallmarks of tumor cells has become a hot topic in oncology. The role of E2 has also become increasingly important in the study of the top ten features of cancer. Although the relationships between cancer hallmarks are complex and often interdependent, we nonetheless attempt to decipher the E2s associated with each specific hallmark. This approach aims to provide illustrative examples for each marker, thereby advancing our understanding of cancer pathogenesis and informing potential therapeutic strategies (Table 1).
E2s in the cancer hallmarks
| Cancer hallmark | E2s | Cancer hallmark | E2s |
|---|---|---|---|
Sustained proliferative signaling | UBE2A [53], UBE2B [54], UBE2C [55, 56], UBE2D3 [57], UBE2E1 [58, 59], UBE2E2 [60], UBE2E3 [61], UBE2F [62], UBE2I [63] UBE2J1 [64, 65], UBE2L3 [66], UBE2M [67], UBE2O [68, 69], UBE2Q 1[70] UBE2Q2 [71], UBE2R1 [72], UBE2T [73, 74], UBE2V1 [75], UBE2V2 [76, 77], UBE2Z[78]. | Inducing angiogenesis | UBE2C [79, 80], UBE2D1 [81], UBE2D3 [82], UBE2E1 [83], UBE2L6 [83], UBE2O [84]. |
Enabling replicative immortality | UBE2D3 [85, 86], UBE2N [87, 88], UBE2T [89]. | Tumor-promoting inflammation | UBE2C [90, 91], UBE2I [92], UBE2L3 [93], UBE2L6 [94], UBE2N [95]. |
Evading growth suppressors | UBE2C [55], UBE2D1 [96], UBE2E3 [97], UBE2F [98], UBE2I [99], UBE2L3 [100], UBE2M [101], UBE2N [102], UBE2Q1 [103], UBE2R1 [104], UBE2S [105]UBE2T [106]. | Inducing invasion and metastasis | UBE2A [107], UBE2B [108], UBE2C [109, 110], UBE2D3 [111], UBE2E2 [112], UBE2F [113], UBE2I [114], UBE2J1 [65], UBE2O [115], UbE2T [73, 74, 116], UBE2V1 [75]. |
Resisting cell death | UBE2A [117], UBE2C [56, 118], UBE2D1 [80, 119], UBE2D2 [120], UBE2D3 [121], UBE2E1 [58], UBE2E3 [61], UBE2F [122], UBE2J1 [64], UBE2L3 [123, 124], UBE2L6 [125], UBE2M [67], UBE2O [126], UBE2S [127], UBE2T [116, 128], UBE2V2 [76, 77]. | Deregulating cellular energetics | UBE2A [129], UBE2C [130-132], UBE2F [62], UBE2I [133], UBE2N [134], UBE2O [135], UBE2Q1 [136], UBE2Q2 [137], UBE2S [138], UBE2T [139]. |
Avoiding immune destruction | UBE2C [140], UBE2D3 [141], UBE2F [113], UBE2I [142], UbE2M [143], UBE2N [144], UBE2R1 [145], UBE2S [146], UBE2T [147], UBE2V2 [148], UBE2W [49]. | Genomic instability and mutation | UBE2A [149], UBE2B [149], UBE2C [150-152], UBE2E2 [60], UBE2I [153, 154], UBE2L3 [93], UBE2N [155], UBE2O [156], UBE2S [157], UBE2T [158]. |
E2s in sustained proliferation signaling
E2s and cell cycle regulators
The cell cycle is governed by a series of checkpoints—namely, the G1/S, G2/M, and spindle assembly checkpoint (SAC)—which collectively ensure orderly progression from one phase to the next [160]. Dysregulation of these checkpoints is critically implicated in the initiation and progression of cancer. Importantly, the cell cycle is extensively modulated by the UPP.
Cyclin B1 serves as a pivotal regulator throughout the cell cycle. It forms a complex with CDK1 to drive mitotic entry. During early M phase, the Cyclin B1/CDK1 complex triggers nuclear envelope breakdown and chromosome condensation, thereby promoting mitosis [161, 162]. Several E2s are intimately linked to Cyclin B1 expression and function, influencing both cell cycle control and tumorigenesis. For example, UBE2A upregulates CCNB1 (which encodes Cyclin B1) via H2B monoubiquitination, modulating Cyclin B1 protein levels (Figure 3B) [163]. Similarly, UBE2C expression shows a strong positive correlation with Cyclin B1. Downregulation of UBE2C reduces cyclin B1 and CDK1 levels by blocking the ERK/AKT signaling pathway, thereby diminishing MPF complex formation and arresting the cell cycle at the G2/M phase. This also inhibits tumorigenesis [164]. In hepatocellular carcinoma, UBE2T exhibits a mechanism similar to that of UBE2C described above. Furthermore, studies have revealed that inhibiting UBE2T leads to downregulation of p21 and p27—both members of the Cip/Kip family of cyclin-dependent kinase inhibitors [165].
Different mechanisms of action of RAD6. (A) RAD6 regulates the transcription of CCND1 likely through affecting the H2B monoubiquitination and H3K4me3 levels at the CCND1 promoter region. (B) RAD6 regulates the transcription of CCNB1 likely through affecting the H2B monoubiquitination. (C) RAD6 interacts physically with HP1α, ubiquitinates HP1α at K154, and further degrades HP1α through the autophagy-lysosome pathway.
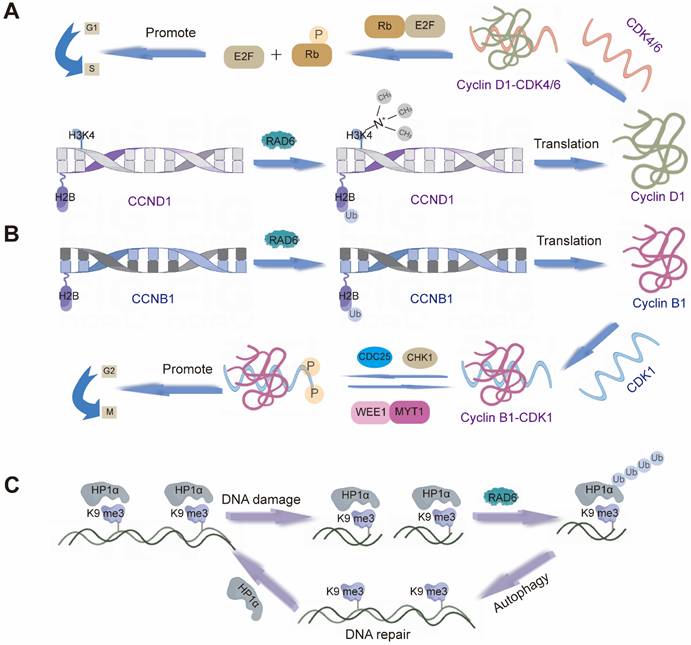
Cyclin D1 is another essential cell cycle regulator, primarily active during the G1 phase. It forms complexes with CDK4 and CDK6, known collectively as Cyclin D1-CDK4/6 complexes [166]. These complexes phosphorylate the retinoblastoma protein (Rb) and other downstream targets to drive G1 progression [167]. Multiple E2s, including UBE2A, UBE2D3, and UBE2M, contribute to the regulation of Cyclin D1. UBE2A, for example, enhances H2B ubiquitination and H3K4me3 levels at the CCND1 promoter, leading to its transcriptional upregulation (Figure 3A) [168]. UBE2D3 (UBE2D3-hTERT-cyclin D1) and UBE2M (Wnt/β-catenin pathway) are believed to influence cyclin D1 stability through distinct mechanisms, thereby participating in cell cycle control [169, 170]. Together, these enzymes fine-tune Cyclin D1 activity and play coordinated roles in cell proliferation. Beyond positive regulators such as Cyclins B1 and D1, negative cell cycle regulators also play crucial roles in maintaining cellular homeostasis by restraining proliferation. E2s are similarly involved in modulating these inhibitory factors. For example, Reports indicate that UBE2D1 mediates the ubiquitination and degradation of the tumor suppressor protein p53 both in vitro and in vivo. In vitro, it interacts with MDM2 to trigger the ubiquitination process of p53. UBE2D1 participates in the ubiquitination of HSP90AB1, which contributes to the stability of p53 and its transport to the cell nucleus [171]. P27kip1 is a member of the Cip/Kip family of CKIs. UBE2I can cooperate with CRM1 to mediate SUMOylation of p27kip1, thereby regulating its nuclear export and influencing cell cycle progression [99]. Moreover, knocking down UBE2M inhibits neddylation modification of Cullin proteins, leading to inactivation of CRL E3s and inducing accumulation of tumor-suppressive CRL substrates (p21, p27, and Wee1). This induces cell cycle arrest and suppresses the malignant phenotype of lung cancer cells [172].
E2s and signaling pathways
Cell growth signaling pathways play crucial roles in maintaining life processes such as cell proliferation, differentiation and survival. Their aberrant activation or inhibition is usually closely associated with the occurrence and development of various diseases, especially cancer [173]. Wnt/β-catenin and NF-κB are some common cellular growth signaling pathways. E2s are directly or indirectly involved in cell proliferation, growth and cancer development by regulating the activity of these signaling pathways.
The Wnt/β-catenin signaling pathway constitutes a fundamental signaling cascade that is essential for numerous physiological processes, including embryonic development, tissue regeneration, and cellular homeostasis [174]. Dysregulation of this pathway, due to genetic or epigenetic alterations, is a well-established driver in various human cancers, such as colorectal, breast, and gastric cancer. A pivotal event in pathway activation is the nuclear accumulation of β-catenin, which results from the functional impairment of the β-catenin destruction complex. E2s are critical regulators of the Wnt/β-catenin pathway. For instance, UBE2S acts as a novel pathway activator by catalyzing a Lys11-linked polyubiquitin chain on β-catenin at Lys19. This specific modification stabilizes β-catenin by antagonizing its Lys48-linked ubiquitination and subsequent proteasomal degradation, which is normally mediated by the destruction complex and β-TrCP [175]. The aberrant activation of this pathway, primarily driven by the nuclear translocation of β-catenin, is a hallmark of many tumors, where it promotes oncogenic gene transcription, cell proliferation, and stem cell self-renewal [162, 176]. The pathological significance of E2s is further underscored by their roles in different cancer contexts: UBE2A and UBE2B influence the pathogenesis of melanoma and breast cancer by modulating the Wnt/β-catenin pathway [177], while UBE2M and UBE2S contribute to its activation in malignancies such as ovarian cancer [178, 179].
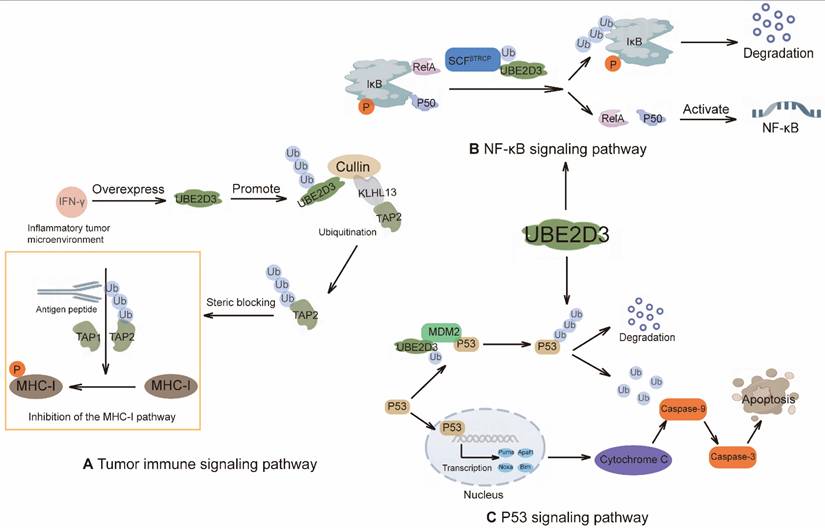
The NF-κB family comprises transcription factors that play critical roles in a range of biological processes, including inflammation, immune response, and apoptosis [180]. Beyond its canonical signaling through NF-κB, this system also exerts effects via interferon regulatory factors [181]. Numerous cellular signaling pathways converge on NF-κB, and ubiquitination plays a crucial role in coordinating these signals to regulate NF-κB activity. In this pathway, the UBE2N-UBE2V1 complex synergistically mediates K63-linked polyubiquitin chain formation with CHIP, enhancing its activity in promoting TACE maturation and thereby blocking TNFα-induced NF-κB signaling [182]. Dysregulation of the NF-κB pathway has been strongly implicated in the development and progression of multiple cancers [183]. E2s such as UBE2D1 and UBE2D3 (Figure 4B) contribute to the precise modulation of this pathway, influencing cancer cell proliferation, survival, and metastatic behavior [184, 185]. Given its central role in oncogenesis, the NF-κB pathway—and its regulatory E2s—represents a promising target for therapeutic intervention in cancer.
Different mechanisms of action of UBE2D3. (A) IFN-γ-driven UBE2D3 upregulation impairs antigen presentation pathways and antitumor immunity in pancreatic cancer. (B) NF-κB/IκB pathway: IκB forms an inhibitory complex with the NF-κB subunits RelA and p50. Phosphorylation of IκB promotes its ubiquitination by the E3 RNF138 in conjunction with UBE2D3, leading to proteasomal degradation of IkB and activation of NF-κB target genes. (C) The role of UBE2D3 in the p53 signaling pathway. The p53 tumor suppressor regulates gene expression and ultimately induces apoptosis.
Role of E2s in replication immortality
One of the characteristics of cancer cells is their abnormal cell proliferation and replication capacity [186]. Normal cells have a limited ability to proliferate under certain conditions, whereas cancer cells can proliferate and divide without restriction to form tumor tissue. Replicative immortality of cancer cells is associated with telomerase activity [187]. Telomerase is an enzymatic protein that protects telomeres at the ends of chromosomes. In normal cells, telomeres are progressively shortened each time the cell divides due to limitations in DNA replication, which eventually leads to cellular senescence and death. However, in cancer cells, telomerase activity is abnormally increased and telomeres are maintained at a constant length, allowing the cancer cells to divide indefinitely and achieve "replicative immortality" [188]. This aberrant expression of telomerase activity is considered to be an important feature of cancer cells and one of the key mechanisms by which cancer cells are able to sustain growth and spread. Therefore, the regulation of telomerase activity has become one of the important research directions in cancer therapy.
In telomere maintenance, ubiquitination modulates telomere length and stability by regulating the stability and function of telomere-associated proteins [189]. In the regulation of telomeric chromatin by ubiquitin, post-translational modifications of histone tails—such as methylation and acetylation—serve as potent mechanisms governing chromatin structure. It has been discovered that RAD6 regulates H3K4-me by adding a ubiquitin molecule to lysine 123 of histone H2B (monoubiquitination of H2B) [190]. As the catalytic subunit of telomerase, the core function of hTERT is to maintain telomere length directly by adding telomeric repeats to chromosome ends in a reverse transcription manner [191]. Studies have reported that UBE2D3 can stimulate hTERT degradation via ubiquitin-dependent proteolysis, leading to reduced telomerase activity and ultimately resulting in telomere shortening [85]. Although the maintenance of telomere length is closely related to ubiquitination, the role of E2s in the regulation of telomerase remains to be further explored.
Role of E2s in evading growth inhibition
Cancer cell evasion of growth inhibition refers to the process whereby tumor cells escape normal regulatory mechanisms and ignore inhibitory signals, resulting in uncontrolled proliferation and dissemination [192]. This phenomenon plays a crucial role at different stages of tumor development, especially during early tumor formation and progression. The mechanism by which cancer cells escape growth inhibition involves multiple aspects, including cell cycle regulation, DNA repair, apoptosis and other biological processes [193]. Cancer cells can evade growth inhibition by regulating the expression of relevant genes, post-translational modification of proteins and activation of signal transduction pathways. These aberrant growth regulatory mechanisms enable cancer cells to continue to proliferate and spread unchecked by external factors, eventually forming malignant tumors [194].
Research indicates that multiple E2s participate in tumor cell escape from growth inhibition by regulating the ubiquitination of key tumor suppressor proteins such as p53 and p27. The stability of p53 is precisely regulated by ubiquitination, and its abnormal degradation is closely associated with tumorigenesis. For instance, significant overexpression of the UBE2T has been observed in various cancers including HCC, CRC, and GC. UBE2T catalyzes K48-or K63-linked ubiquitination of p53, thereby promoting its degradation or altering its function. This ultimately disrupts p53-dependent downstream signaling pathways, weakening its growth-inhibitory and pro-apoptotic effects [116]. Conversely, the ubiquitin-mediated degradation of the cyclin-dependent kinase inhibitor p27 is a critical step in releasing cell cycle arrest and driving cells into the DNA replication phase. Research indicates that UBE2S interacts with the scaffold protein TRIM28 within the nucleus, synergistically enhancing p27's ubiquitination levels and accelerating its degradation [45]. The loss of p27 removes inhibition on the cyclin-kinase complex, propelling cell cycle progression and enabling cells to bypass normal proliferation surveillance.
The abnormally high expression of these E2s disrupts the homeostasis of core tumor suppressor proteins like p53 and p27, thereby undermining critical biological processes such as cell cycle checkpoints, DNA damage response, and apoptosis pathways. This creates favorable conditions for the uncontrolled proliferation and dissemination of tumor cells.
Role of E2s in the regulation of cell death
The UPP plays a crucial role in the regulation of apoptosis, a process essential for maintaining cellular homeostasis under various dysregulated conditions [184]. Key apoptotic regulators include the BCL-2 protein family, IAPs, and the IKK complex [195]. Apoptosis primarily proceeds through two major pathways: the extrinsic and the intrinsic routes. The extrinsic pathway is initiated by extracellular signals such as death ligands (TNF, FASL), cytokines, cytotoxic stimuli, or alterations in the extracellular matrix [196]. These signals are transduced via cell surface receptors, leading to caspase activation and the execution of apoptosis [197]. In contrast, the intrinsic pathway is triggered by intracellular stressors, including DNA damage, metabolic dysfunction, or cell cycle dysregulation. These stimuli induce mitochondrial outer membrane permeabilization, resulting in the release of pro-apoptotic factors such as cytochrome C and BCL-2 family proteins.
In MM, the transcription factor c-Maf is frequently overexpressed in more than 50% of cell lines and patient samples, primarily due to chromosomal translocations and other mechanisms [198]. Notably, UBE2O has been identified as a key regulator that induces K48-linked polyubiquitination of c-Maf, suppresses its transcriptional activity, and thereby promotes MM cell apoptosis while inhibiting tumor growth [199]. As a central tumor suppressor, p53 regulates apoptosis through the transcriptional control of genes such as PUMA, NOXA, BIM, BAX, BCL2, FAS, and IGFBP3, and can also trigger apoptosis via death receptor pathways like those involving TNF and Fas [200]. For instance, UBE2D3 cooperates with MDM2 to conjugate K48-linked polyubiquitin chains onto p53, triggering its proteasomal degradation. This modulates the diverse activities of p53, including its ability to promote apoptosis [201]. Furthermore, inhibiting UBE2J1 can regulate the PI3K/AKT and MDM2/p53 signaling pathways to promote apoptosis in endometrial cancer [202].
BAX, a key apoptotic effector, undergoes conformational activation and mitochondrial translocation upon cellular stress, forming pores that facilitate cytochrome c release and apoptosome formation [203]. UBE2L3 promotes the ubiquitin-mediated proteasomal degradation of GSK3β, the loss of GSK3β blocks p65 activation. This leads to the upregulation of p65 target proteins such as Bax, ultimately influencing apoptosis in HCC [124]. Similarly, the pro-apoptotic protein NOXA, which enhances cellular sensitivity to apoptosis, is regulated by UBE2F-mediated ubiquitination and degradation. UBE2F pairs with SAG/RBX2 to mediate neddylation of CUL5, thereby activating the CRL5 E3 complex. Activated CRL5 then conjugates K11-linked ubiquitin chains to NOXA, targeting it for degradation and consequently inhibiting apoptosis to enhance cell survival [204]. Unlike other BCL-2 family members, NOXA expression is subject to multi-level regulation, including transcriptional control, post-translational modification, and protein degradation [205]. Its overexpression promotes apoptosis in numerous cancer types, highlighting its potential as a therapeutic target in oncology [206].
Role of E2s in immune escape
Immune escape is a key process in tumor development that involves tumor cells using various mechanisms to avoid detection and clearance by the immune system, thereby promoting tumor growth, metastasis and treatment resistance [207]. Multiple E2s, including UBE2D3, UBE2I, UBE2N, UBE2S, UBE2L6 and UBE2F are implicated in this process. These E2s may influence cells' ability to evade the immune system by regulating antigen presentation, immune checkpoint/co-stimulatory molecules, cytokine/chemokine signaling pathways, and the shaping of an immunosuppressive microenvironment.
Our recent studies have identified UBE2D3 as a key driver of inflammation-dependent immune escape in pancreatic cancer. The underlying mechanism involves the inflammatory microenvironment inducing UBE2D3 overexpression in tumor cells, which then associates with the E3 ligase KLHL13 to mediate K63-linked polyubiquitination of lysine 245 on the transporter associated with antigen processing 2 (TAP2). This modification creates steric hindrance that impairs the peptide-transport function of TAP2, thereby blocking the antigen presentation pathway and facilitating immune evasion by tumor cells [141] (Figure 4A).
Immune checkpoints refer to a class of immune inhibitory molecules expressed on immune cells, which fine-tune the degree of immune activation and prevent the occurrence of autoimmunity. The expression of immune checkpoint molecules is precisely regulated by E2s. In prostate cancer, UBE2I within macrophages mediates SUMOylation modification at lysine 350 of STAT4, promoting its ubiquitination and degradation. This subsequently inhibits macrophage polarization towards the M1 phenotype via the JAK/STAT signaling pathway. Application of the UBE2I inhibitor 2-D08 enhances the anti-tumor function of tumor-associated macrophages, stimulates the proliferation and activation of CD8⁺ T cells, and upregulates PD-1 expression, demonstrating synergistic potential when combined with immune checkpoint blockade therapy [142]. In liver cancer, miR-122-3p downregulates PD-L1 expression and enhances anti-tumor immune responses by targeting UBE2I and inhibiting the SUMOylation modification of the NF-κB signaling pathway [303]. Conversely, inhibition of UBE2N in macrophages reduces K63-linked polyubiquitination, leading to AKT activation and upregulation of PD-L1 expression, thereby promoting tumor progression [144].
Beyond immune checkpoint blockade, targeting cytokine/chemokine signaling pathways represents another promising strategy in cancer immunotherapy. The IL-15 signal is crucial for maintaining the function of cytotoxic lymphocytes. UBE2F inhibits NK cell function and suppresses the secretion of pro-inflammatory cytokines by mediating the Neddylation modification of CUL5, which activates the CRL5 complex. This activated complex subsequently targets the IL-15 receptor for proteasomal degradation [113]. IFN-I is also one of the most critical categories of cytokines in the immune system, playing a central role in initiating and coordinating early antiviral innate immune responses. Within the IFN-I pathway, UBE2S negatively regulates this signaling axis by recruiting USP15 to remove the K63-linked polyubiquitin chains from TBK1 [208]. Furthermore, RSK1-mediated phosphorylation of UBE2L6 alters its substrate preference, enabling it, in concert with RNF19A, to drive cGAS ubiquitination. This process consequently inhibits cGAS-STING pathway-mediated IFN-I production and intrinsic anti-tumor immunity [209].
In shaping the immunosuppressive tumor microenvironment, UBE2S also plays a significant role. The expression level of UBE2S shows a positive correlation with the infiltration of M2-polarized tumor-associated macrophages, suggesting its potential as a biomarker for predicting responses to immunotherapy [210]. Mechanistically, research has revealed that UBE2S recruits USP10 through the N-terminal domain of USP10. Together, they regulate the K48-linked deubiquitination of the GLUT1 protein, thereby stabilizing GLUT1 and enhancing glycolytic activity. This metabolic reprogramming leads to lactate accumulation, which in turn induces the differentiation of M2-polarized macrophages and the secretion of TGF-β1. These changes further promote the transformation of fibroblasts into myofibroblasts, accelerating the process of tissue fibrosis [211].
Although current research on the specific mechanisms by which E2s affect immune escape remains insufficient, these E2s likely play a significant role in the immune escape process of tumor cells by regulating the expression of immune-related genes, influencing immune-related signaling pathways, and promoting tumor growth and metastasis.
Role of E2s in angiogenesis
Normally, angiogenesis is a temporary process that occurs only under specific physiological and pathological conditions, such as wound healing and endometrial proliferation. However, cancer cells can manipulate and promote angiogenesis through different mechanisms to meet their nutritional needs for continued growth and spread [212]. In angiogenesis, E2s play a key role.
Vascular endothelial growth factor receptor 2 (VEGFR2) serves as a central regulator of endothelial cell function and angiogenesis. During angiogenesis, the E2 ubiquitin-conjugating enzymes UBE2D1 and UBE2D2 modulate VEGFR2 ubiquitination and fine-tune endothelial responses to VEGF-A signaling. Inhibition of either UBE2D1 or UBE2D2 elevates the steady-state level of VEGFR2, thereby potentiating VEGF-A-stimulated signal transduction and amplifying the activation of downstream pathways, including the classical MAPK cascade, phospholipase Cγ1, and the AKT pathway [81].
Beyond VEGFR2 regulation, UBE2D1 also influences angiogenesis—particularly in muscular contexts—by targeting hypoxia-inducible factor-1α (HIF-1α), a master cellular oxygen sensor and key transcriptional regulator of angiogenesis. Under normoxic conditions, HIF-1α undergoes proline hydroxylation at residues 402 and/or 564 catalyzed by prolyl hydroxylase domain proteins (PHDs) 1-4. This modification promotes its recognition by the Von Hippel-Lindau tumor suppressor (VHL) and subsequent ubiquitination facilitated by UBE2D1, leading to proteasomal degradation and maintenance of low basal levels. In contrast, under hypoxic conditions, PHD and VHL activities are suppressed, resulting in HIF-1α stabilization, transcriptional activation, and the promotion of angiogenesis, glycolysis, and cell survival [213].
Furthermore, the expression of UBE2C is closely linked to the angiogenic phenotype. Knockdown of UBE2C downregulates key pro-angiogenic factors such as VEGF and MMP-9. Its positive correlation with microvessel density (MVD) and vasculogenic mimicry (VM) has been established in both non-small cell lung cancer and breast cancer, supporting its role in tumor-associated angiogenesis [80, 200, 214].
Tumor necrosis factor receptor 1 (TNFR-1) also represents a key regulator of angiogenic activity. In the mechanism governing angiogenesis regulation, UBE2N specifically catalyzes K63-linked polyubiquitination—a modification distinct from the canonical K48-linked ubiquitination that typically targets proteins for degradation [215]. Within this mechanism, UBE2N forms an immune complex with the E3 ligase RNF213, termed the RNF213-UBE2N complex. RNF213 may mediate K63-linked polyubiquitination of TNFR-1 ligands through this complex [216].
In summary, these studies demonstrate that specific E2s critically regulate angiogenesis through distinct mechanisms, such as modulating the stability of key receptors, the expression of angiogenic factors, and the dynamics of specific ubiquitin chain linkages.
Role of E2s in cellular energy metabolism
Tumor cells exhibit energy metabolism patterns markedly different from normal cells, a characteristic known as metabolic reprogramming, which is a core hallmark of their malignant phenotype [217]. Even under adequate oxygen supply, tumor cells preferentially utilize the glycolytic pathway for energy production, a phenomenon termed the Warburg effect. This allows for the rapid generation of ATP and lactate, providing the metabolic foundation for their sustained proliferation and invasion.
E2s play multi-layered regulatory roles in this metabolic rewiring. UBE2C can interact with the epidermal growth factor receptor (EGFR) and enhance its stability, leading to sustained activation of the downstream PI3K-AKT signaling axis. This UBE2C-EGFR-PI3K/AKT regulatory network serves as a critical hub driving metabolic reprogramming and can significantly promote glycolytic flux [130]. The activated PI3K-AKT pathway further reinforces the Warburg effect, accelerating the conversion of glucose to lactate and the rapid production of ATP, thereby supplying the necessary bioenergy and biosynthetic precursors for tumor cell proliferation [218].
Beyond the aforementioned pathway, UBE2S can directly catalyze K11-linked polyubiquitination of the VHL protein at lysine residues 171 and 196 in an E3-independent manner, thereby promoting its proteasomal degradation. The loss of VHL indirectly stabilizes the hypoxia-inducible factor HIF-1α, ultimately upregulating the expression of glycolysis-related genes in an HIF-1α-dependent manner [138].
Other E2 members also participate in metabolic remodeling through distinct mechanisms. For instance, the circPDK1/BIN1/UBE2O ternary complex reduces BIN1 stability via ubiquitination, thereby enhancing c-Myc transcriptional activity and driving glycolysis in pancreatic cancer [135]. Meanwhile, UBE2Q2 binds to cIAP1 and mediates K63-linked ubiquitination of receptor-interacting protein 1 (RIP1), activating the NF-κB pathway, which upregulates HIF1α transcription and enhances glycolysis [137].
Furthermore, tumor cells can utilize alternative carbon sources such as fatty acids and amino acids for energy supply and biosynthesis, with E2s also playing regulatory roles in these adaptive metabolic processes. This metabolic plasticity enables tumor cells to maintain a survival advantage in diverse microenvironments, thereby acquiring greater potential for progression and treatment resistance.
Role of E2s in invasion and metastasis
Cancer invasion and metastasis are one of the most critical steps in cancer development and one of the main causes of treatment failure and death. During tumor progression, cancer cells can break through the boundaries of the original tumor, invade surrounding tissues and enter the bloodstream or lymphatic system, eventually forming metastases in other parts of the body [219]. This process involves the regulation of complex signaling pathways and molecular mechanisms, among which E2s, as one of the key regulators, play an important role in cancer invasion and metastasis.
Vimentin, a type III intermediate filament protein, serves as a key regulator of EMT, a process critical for tumor invasion and metastasis. UBE2C drives the invasion and metastasis of follicular thyroid carcinoma by mediating K29 site-specific ubiquitination of vimentin, thereby regulating the EMT machinery [220]. In addition, a conserved miRNA, miR-525-5p, was found to target UBE2C and subsequently regulate the expression of ZEB1/2 [221]. Mechanistically, supplementation of UBE2C partially rescues the inhibitory effect of miR-525-5p on cellular invasion. UBE2D1 primarily exerts its pro-metastatic function via the TGF-β/SMAD4 pathway. Knockdown of UBE2D1 reduces the ubiquitination level of SMAD4, thereby attenuating TGF-β/SMAD4 signaling. This leads to downregulation of EMT markers and consequently inhibits cell migration and metastasis in both in vitro and in vivo models [222].
There are also a number of E2s, such as UBE2T and UBE2J1, that can affect cellular function and thus cancer cell invasion and metastasis through different mechanisms. For example, overexpression of UBE2T may promote prostate cancer cell proliferation and metastasis by activating AKT/GSK3β/β-linker pathway [223]. Functionally, UBE2J1 could inhibit the proliferation and metastasis of CRC cells in vitro and in vivo. The UBE2J1-TRIM25 complex induces ubiquitination and degradation of RPS3 at the K214 residue. Downregulation of RPS3 inhibits NF-κB translocation into the nucleus and therefore inactivates the NF-κB signaling pathway [65].
Based on the integrated analysis of studies in this section, it can be concluded that E2s are not only involved in conventional intracellular protein synthesis and degradation pathways, but also extensively participate in multiple biological processes of tumor cells—including growth, migration, invasion, death, metabolism, and immune microenvironment remodeling—through positive or negative regulation of key signaling pathways and cytokines.
E2s in enabling characteristics of cancer
E2s in tumor-promoting inflammatory signaling
In recent years, inflammation has been increasingly recognized as a major hallmark of cancer [224]. Aberrant signaling cascades during inflammation promote chronic inflammation, contributing to tumorigenesis [225]. E2s play a critical role in regulating inflammatory signaling pathways, and the loss or overexpression of E2s may lead to aberrant inflammatory signaling in specific contexts.
UBE2N influences cancer-promoting inflammatory signals by regulating the TRAF6-mediated NF-κB signaling pathway, a signaling cascade frequently hijacked in tumor-promoting inflammation [226]. The NF-κB signaling pathway plays diverse roles in immunity and development and is frequently hijacked to regulate tumor-promoting inflammation. In TLR and RANK signaling pathways, the E3 ligase TRAF6 forms a functional complex with its dedicated E2 partner, UBE2N [227]. This TRAF6-UBE2N complex specifically catalyzes K63-linked polyubiquitination, which is required for the subsequent activation of the IKK complex. IKK activation then drives NF-κB signaling, leading to nuclear translocation and pro-inflammatory gene expression [228]. Additionally, similar to NF-κB, STAT3 acts as a transcription factor regulating anti-inflammatory responses, and it can limit RANK - and TLR4-mediated signaling by suppressing expression of UBE2N [229]. Studies have also found that loss of UBE2N in keratinocytes leads to skin inflammation, increased epithelial cell proliferation, and significant infiltration of immune cells, particularly neutrophils and macrophages. This inflammatory response can be alleviated by affecting the TLR/IL-1 signaling pathway-mediated molecules IRAK1/4 [230]. Therefore, UBE2N may also participate in promoting cancer-related inflammatory responses by modulating the TLR/IL-1 signaling pathway.
Other E2s associated with inflammatory diseases is UBE2D2, which plays an important role in the inflammatory process of atherosclerosis. Under oxidized ox-LDL stimulation, intracellular expression of miR-30b-5p is upregulated. miR-30b-5p binds to UBE2D2, reducing its ubiquitin conjugation ability, leading to increased stability of KAT2B. The upregulation of KAT2B promotes the acetylation of HMGB1, resulting in the dissociation of HMGB1 from the deacetylase SIRT1, ultimately leading to the release of HMGB1 [231, 232]. The release of HMGB1 further triggers the inflammatory response and promotes polarization and migration of macrophages [232]. Therefore, UBE2D2 participates in the regulation of the inflammatory response by modulating the ubiquitination and acetylation levels of KAT2B and HMGB1 during inflammation. Given the similarity between the inflammatory signals of tumors and the inflammatory process of atherosclerosis, it is speculated that UBE2D2 may play a role in certain types of tumors.
E2s convert chronic, uncontrolled inflammatory responses into drivers supporting malignant tumor progression through precise ubiquitination codes. Future research should no longer view E2s in isolation within a single pathway, but rather focus on their networked functions throughout the entire tumor microenvironment system. By deciphering this intricate network, we may uncover the “master switch” controlling tumor-promoting inflammation, paving the way for novel therapies that reprogram the tumor microenvironment and fundamentally reshape the landscape of cancer treatment.
E2s in genomic instability and mutation
Genomic instability typically refers to the presence of various DNA alterations, ranging from single nucleotide changes (such as base substitutions, deletions, and insertions) to chromosomal rearrangements (gains or losses of chromosomal segments or entire chromosomes [233]. Loss of genomic stability can lead to the early onset or accelerated progression of degenerative diseases, including premature aging and cancer. DNA serves as the template for fundamental processes like replication and transcription; therefore, maintaining its integrity is crucial for cell survival. DNA repair is essential for maintaining genomic integrity for cell survival, and failure to repair DNA damage can lead to genomic instability, cancer, or even cell death [234]. The loss or mutation of DNA repair functions is a fundamental cause of genomic instability in hereditary cancers [235]. Genomic instability is closely associated with the ubiquitination process, in which E2s such as RAD6, UBE2D3, UBE2I, UBE2L3, and UBE2N play crucial roles.
For example, RAD6 regulates its function in DNA repair by modifying proliferating cell nuclear antigen (PCNA), a key coordinator of DNA replication and repair processes, which participates in various DNA repair pathways through ubiquitination and SUMOylation [236]. Besides, RAD6 plays a key role in repairing UV-induced DNA damage, RAD6 physically interacts with HP1α and ubiquitinates HP1α at residue K154, thereby promoting HP1α degradation through the autophagy pathway and ultimately leading to an open chromatin structure that promotes efficient HR DSB repair. In addition, bioinformatics studies have shown that the expression of RAD6 and HP1α has an inverse relationship and is related to the survival rate of patients [237, 238] (Figure 3C). Multiple E2s are critically involved in maintaining genomic integrity through distinct DNA damage response and repair pathway. For instance, downregulation of UBE2D3 enhances the DNA damage repair pathway, leading to increased expression of key proteins such as ATM, ATR, p-ATM, cyclin D1, and CHK1. Concurrently, it results in the suppression of DNA damage and cell cycle regulators, including γH2AX, CDC25A, and CDC25C [169]. ZHIYUAN SHEN et al. found that UBE2I interacts specifically with DNA repair-related proteins such as RAD52, RAD51, p53, and UBL1 in a yeast two-hybrid system [6]. This specific interaction may affect DNA repair pathways, thereby influencing genomic stability. UBE2L3 regulates the role of 53BP1 in the repair of DSBs by promoting the ubiquitination and degradation of 53BP1 [239], a key factor in DNA DSBs By regulating 53BP1 abundance, UBE2L3 modulates the cellular response to replication stress, thereby affecting the choice of DSB repair pathways, which may affect the cell's DNA repair capacity and genomic stability. UBE2N redistributes to the nucleus upon DNA damage and facilitates the assembly of complexes within the RAD6 pathway of DNA repair, preventing genomic instability and cancer. When cells are subjected to DNA damage, UBE2N forms heterodimers with non-catalytic E2-like partner proteins Mms2 and Uev1A, which play a role in the RAD6 pathway of DNA repair [240], similar to UBE2S and UBE2W. UBE2S binds to Ku70 and affects non-homologous end-joining (NHEJ)-mediated DNA repair processes through this interaction, which may affect the accuracy and efficiency of DNA repair [241]. UBE2W interacts with DNA repair-related proteins such as BRCA1 and BRCA2, which are involved in the repair of DNA double-strand breaks [49].
Chromosomal instability, which arises from sustained errors in chromosome segregation during mitosis, is the most common form of genomic instability [242]. Excessive induction of random chromosomal instability can trigger detrimental chromosomal patterns in cells [243], which may promote tumorigenesis. This process is also regulated by ubiquitination process. UBE2C participates in the activation of the APC/C, an important regulator of cell mitosis [169]. Overexpression of UBE2C may induce premature activation of APC/C, resulting in erroneous chromosome segregation and mitotic progression, which directly promotes chromosomal instability. Similarly, downregulation of UBE2D3 leads to increased telomerase activity and upregulation of various proteins in the hTERT and shelterin complexes, such as TRF1, TRF2, POT1, and RAP1. These proteins play a crucial role in protecting telomeres and maintaining chromosomal stability. Thus precise regulation of distinct E2s is critical for maintaining chromosomal stability through multiple mechanisms.
Targeting E2s to treat cancer
E2 occupies a pivotal position in the ubiquitin-proteasome system, directly determining the fate of substrate proteins and serving as a key regulator of cell cycle progression [53], DNA repair, and signal transduction [244]. Therapeutic strategies targeting E2 aim to precisely intercept multiple oncogenic pathways upstream by specifically inhibiting E2 members abnormally activated in cancer, offering a novel approach for achieving highly effective and low-toxicity tumor treatment. E2s in cancer are context-dependent, arising from the specific pathways they regulate. E2s are involved in a variety of physiological functions within cells, and their impact on cancer is often complex. For instance, specific E2 and the ubiquitin ligase Parkin protein mediate the mitophagy pathway in cells, while E2 containing the baculovirus IAP repeat sequence may inhibit the expression of autophagic flux [245]. Many E2s exert multiple, sometimes opposing, roles in different cellular environments and pathways (Figure 5). So, is there a perfect target in E2 that deserves to be the focus of cancer drug discovery efforts? This question is not easy to answer. The current literature predominantly focuses on a subset of E2s that are most relevant to cancer and often touted as valuable therapeutic targets. These tend to be well-studied, widely expressed enzymes for which research tools are readily available. Consequently, this focus may have obscured the potential roles of low-abundance, specialized E2s that are dysregulated in specific cancer contexts. Overall, the ratio of charged to uncharged E2 and the concentration of free ubiquitin determine whether the OTU deubiquitinase, ubiquitin aldehyde-binding 1-E2 complex can act as a deubiquitinase or polyubiquitination inhibitor [246]. The ubiquitin system plays a pivotal and multifaceted role, representing a rich source of clinical therapeutic targets. Preclinical evidence demonstrates the feasibility of using small molecule inhibitors of E2s to attenuate the degradation of many tumor suppressor substrates (Table 2), inhibit cell proliferation in vivo, and inhibit tumor growth in vivo.
E2 targets in cancer. Ubiquitin-conjugating enzymes (E2s) play multifaceted roles in cancer initiation and progression, with their functions highly dependent on the cellular environment. Most E2s exhibit dual tumor-suppressor or oncogenic activities under specific conditions. Although understanding of E2 mechanisms in tumors remains nascent, several members have emerged as key preclinical targets due to their unique properties and capacity to drive tumorigenesis by regulating multiple cancer hallmarks.
Inhibitors targeting E2s
| Name | Chemical structure | Molecular formula | Inhibitor category | Origin | Target | Mechanism of action | In vitro inhibition rate | Tumor inhibition rate | Cancer Type | Refs |
|---|---|---|---|---|---|---|---|---|---|---|
| TZ8 | 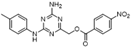 | C18H16N6O4 | Non-covalent inhibitor | Synthesis | UBE2B | Inhibits H2A ubiquitination. | IC50:25μM (MDA-MB-231) | - | TNBC | [283] |
| TZ9 | 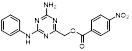 | C17H14N6O4 | Non-covalent inhibitor | Synthesis | UBE2B | Inhibits H2A ubiquitination. | IC50: 6μML (MDA-MB-231) | - | TNBC | [283] |
| New triazines | 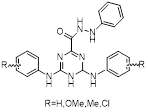 | R=H: C17H19N7OR=OMe: C19H23N7O3R=Me: C19H23N7OR=Cl: C17H17Cl2N7O | Non-covalent inhibitor | Based on TZ8 / TZ9 synthesis | UBE2B | Inhibits H2A ubiquitination. | IC50: 3.3 to 22μM (OV90, A2780, H1299, A549, MCF-7, MDA-MB231 and HT29) | - | OC, BC, CRC | [284] |
| EN450 | 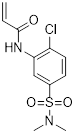 | C11H13ClN2O3S | Allosteric inhibitor | Synthesis | UBE2D1 | Covalently binds UBE2D and exhibits molecular glue interactions with NFKB1. | Approximately 75% inhibition rate at 5μM (HAP1) | - | CML | [285] |
| PC3-15 | 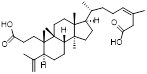 | C32H52O4 | PPI inhibitor | Natural product compounds | UBE2D2 | Binds UBE2D2 and inhibits p62 ubiquitination | EC50: 13.5μM (MDA-MB-468) | MDA-MB-468 and HCC1806 xenografts: Lapatinib & PC3-15: 50 mg/kg (approximately 50%) | TNBC | [286] |
| DHPO | 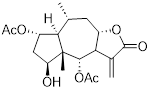 | C10H18O3 | Non-covalent inhibitor | Natural product compounds | UBE2D3 | Binds to UBE2D3 and inhibits NF-κB activation | IC50: 2.5 to 8.5μM (CFPAC1, HPAC, BxPC3, Panc1, SW1990, Capan2, AsPC1, and HPNE) | Panc1 in situ tumor model: 10mg/kg (80.67%) SW1990 in situ tumor model: 10mg/kg (78.74%) | PC | [222] |
| HA-9104 | 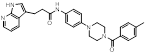 | C28H27N5O2 | Non-covalent inhibitor | Synthesis | UBE2F | Impairs UBE2F-NEDD8 thioester formation and inhibits Cullin-5 neddylation | IC50: 5.26μM (H1650) IC50: 1.7625μM (H2170) IC50: 4.377μM (H358) | H1650 xenograft models: 30 mg/kg (approximately 30%) | LC, PC | [287] |
| CW3 | 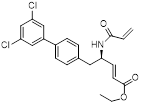 | C22H21Cl2NO3 | PPI inhibitor | Synthesis | UBE2G2 | Forms a covalent bond with the thiol group of Cys48 of UBE2G2. | >50% inhibition rate in 24 cell lines (10μM) | - | - | [288] |
| 2-D08 | 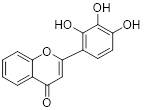 | C15H10O5 | Non-covalent inhibitor | Synthesis | UBE2I | Induces ROS accumulation-mediated intrinsic apoptosis possibly through deSUMOylation of NOX2. | IC50: 18μM (MOLM13) IC50: 15μM (ML2) | - | CML | [289] |
| NSC697923 | 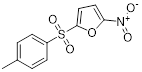 | C11H9NO5S | PPI inhibitor | Synthesis | UBE2N | Inhibits UBE2N-Ub conjugate formation, activates p53 and JNK pathways. Inhibits UBE2N-Ub conjugate formation, activates the MEK/FRA1/SOX10 signaling cascade. | IC50: 1 to 5μM (IMR32, NGP, NB19, CHLA-255, SK-N-AS, and, SH-SY5Y) IC50: 4 to 8μM (A375, A2058, and B16) | SH-SY5Y and NGP orthotopic model: 5 mg/kg (approximately 85%) A375 xenograft model: 5 mg/kg (approximately 75%) | NB, MM | [269, 290] |
| UC-764864 | 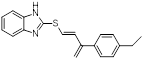 | C19H18N2OS | Covalent inhibitor | Synthesis | UBE2N | Interferes with UBE2N to eliminate the function of hematopoietic stem/progenitor cells in leukemia. | IC50:4.3 to 6.7 µM (HL-60, NOMO-1, Kasumi-1, MV4-11, and MDSL) | - | AML | [263] |
| ATO | 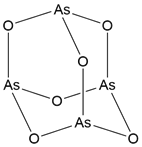 | As2O3 | Covalent inhibitor | Natural product compounds | UBE2O | Inhibits the ubiquitination and degradation of AMPKα2, suppressing the activation of the mTOR-HIF1α pathway. | - | UBE2O+/+ orthotopic tumor model: 2.5 mg/kg (approximately 75%) | BC, PC | [44] |
| CC0651 | 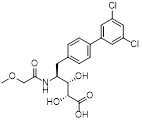 | C20H21Cl2NO6 | Allosteric inhibitor | Synthesis | UBE2R1 | Stabilizes UBE2R1-Ub interaction,Impairs Ub transfer. | IC50: 1.72 μM (PC-3 and HCT116) | - | PC, CRC | [277] |
| CU2 | 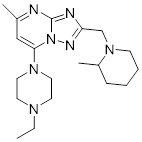 | C19H31N7 | PPI inhibitor | Synthesis | UBE2T | Blocks UBE2T/FANCL-mediated monoubiquitination of FANCD2, thereby enhancing cellular sensitivity to the DNA-crosslinking agent carboplatin. | - | - | OC | [291] |
| M435-1279 | 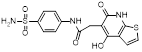 | C18H17N3O5S2 | Non-covalent inhibitor | Synthesis | UBE2T | Inhibits the excessive activation of the Wnt/β-catenin signaling pathway by blocking UBE2T-media-ted degradation of RACK1. | IC50: approximately 10 μM (HGC27, MKN45, and AGS) | - | GC | [292] |
Structure determines function. To develop E2-targeting inhibitors, it is essential to first understand the structural characteristics of E2s. The UBC domain is the core catalytic unit of E2, responsible for generating the E2-Ub conjugate. This domain typically adopts an α/β-fold structure, consisting of four α-helices and four β-strands, with key loop regions constituting part of the E3-binding site (L1 and L2 loops) and the active site (L3 loop) [11]. The highly variable, flexible L1 and L2 loops, extending from the C-terminus to strand 1 and strand 3, are responsible for binding specific E3 ligase [247]. The L1 loop in E2 is crucial for this interaction: its sixth residue acts as an interfacial hotspot and is indispensable for E2-E3 interaction. Furthermore, residues at other positions on the L1 loop participate in regulating the specificity of E2-E3 interactions. For example, HECT-type E3 ligases can contact the second residue of the L1 loop, while RING-finger-type E3 ligases do not exhibit this interaction pattern [248]. A highly conserved loop anchors H2 to strand 4 and, together with H3, forms a shallow residue groove that accommodates the active site cysteine [247]. Structural elements surrounding the active site Cys are involved in processing and activating Ub/Ubl [11, 249]. The catalytic groove also houses other conserved residues, the most important of which is the three-residue HPN motif located upstream of the active site cysteine residue. The secondary structural elements of these enzymatic catalytic domains are highly conserved, suggesting the existence of a universal mechanism promoting enzyme function [250].
Current research on the therapeutic potential of targeting E2s remains incomplete, with more comprehensive studies available only for specific E2s such as UBE2T, UBE2C, and UBE2N. UBE2T possesses a typical UBC domain along with an extended, less conserved, and predominantly disordered C-terminal region [250]. Structural analysis reveals that its surface lacks obvious deep pockets or stable small-molecule binding sites, which may be one reason for the low success rate in drug development targeting UBE2T. The catalytic activity of UBE2T depends on cysteine 86 (Cys86) [251]. Mutation of this residue (C86) completely blocks its ubiquitin transfer function. Additionally, mutation of key lysine sites on its downstream target proteins (K119/120 of H2AX [158], K8/14 of AKT [252]) similarly hinders the completion of ubiquitination.
Functionally, UBE2T can cooperate with up to 15 different E3s [253], including Mule [254], INF8 [158], FANCL [255], among others. Besides the conserved UBC domain, UBE2T also contains multiple flexible regions that may undergo conformational changes when binding to different E3s; its N-terminal helix exhibits significant conformational rearrangement, swinging from a position distant from the active site to a position closer to FANCL, forming a tighter, more specific binding interface [256]. Furthermore, UBE2T itself is subject to ubiquitin modification, containing multiple lysine sites (including Lys28, 48, 91, and 192, etc.); this self-ubiquitination affects protein degradation by regulating UBE2T stability [251]. UBE2C possesses a typical UBC domain along with an N-terminal extension of 28 residues. The active site cysteine responsible for catalyzing ubiquitin-adduct formation, Cys114, is located between β4 and a 310 helix. The three-residue-per-turn structure of the 310 helix positions its positively charged Lys119 residue adjacent to the active site Cys114, together facilitating ubiquitin-adduct formation [257]. Four β-turns (β1-β4) provide a contact surface for the active site Cys114 [258]. Additionally, the ubiquitination and degradation of UBE2C itself depend on Cys114 and a D-box-like motif located at residues 129-132 [257, 259]. UBE2C works closely with the E3 ligase APC/C, together forming a key molecular machinery regulating cell mitosis progression and exit [260]. Notably, while the N-terminal extension of UBE2C is not essential for ubiquitin-adduct formation, it participates in regulating APC/C activity as part of an inhibitory mechanism [257]. Furthermore, the QNP motif (Gln4, Asn5, and Pro8) within its N-terminal extension exerts a negative regulatory effect on APC/C activity through interaction with APC/C [261]. In drug development targeting UBE2C, one of the main challenges stems from the three-dimensional structural features of E2s-their catalytic sites are located in relatively shallow and conserved regions, making it difficult for small-molecule inhibitors to achieve high-affinity, specific binding [262]. UBE2N has Cys87 as its catalytic active center [263]. The morphology of the active site near this residue is regulated by conformational changes in a flexible loop composed of amino acids 114-124. This loop can adopt different conformations, thereby altering the volume and shape of the active site cavity and significantly affecting the morphology and distribution of potentially druggable binding pockets around Cys87 [264]. In co-crystal structures with covalent inhibitors, three druggable pockets have been identified in UBE2N [265]. The position, shape, and size of these pockets vary with the arrangement of surface amino acids. In UBE2N, the transition of its active site loop from an inactive to an active state may be partially triggered by Asn123 - a residue unique to UBE2N. In the inactive loop conformation, Asn123 is buried within the protein, forming a hydrogen bond network with the backbone carbonyl oxygens of His77, Pro78, and Val80. In the active conformation, Asn123 rotates to the surface, forming a hydrogen bond with the backbone amide of the attacking acceptor ubiquitin's Lys63, thereby driving the structural rearrangement of the entire loop [266, 267]. UBE2N interacts with various proteins, including not only E3s but also essential E2 cofactors-the nuclear UBE2V2 and the cytoplasmic UBE2V1. Notably, the binding interfaces between UBE2N and these two cofactors are identical. UBE2V2 and UBE2V1 are highly similar in structure and sequence. Their three-dimensional structures are highly conserved, with the few non-conserved residues located outside the binding interface with UBE2N. Therefore, inhibitors targeting the interaction between UBE2N and UBE2V2 are likely to also affect the binding between UBE2N and UBE2V1 [268, 269].
Based on our reading and analysis of the relevant literature, a handful of E2s stand out for their broad and well-established roles in promoting multi-pathway cancer. Many other E2s commonly cited as cancer targets may have both tumor-promoting and tumor-suppressive effects, depending on the environment, providing a higher standard for generating safe small molecule drugs.
Currently reported E2 inhibitors primarily function through three modes of action: binding to the active site, allosteric sites, or protein-protein interaction interfaces [270]. Among these, active site inhibitors can be further categorized into covalent and non-covalent types: covalent inhibitors exert their effects by forming irreversible or reversible covalent bonds with the catalytic cysteine residue, whereas non-covalent inhibitors reversibly bind to the target via hydrogen bonds, hydrophobic interactions, van der Waals forces, etc. Covalent inhibitors offer advantages such as prolonged residence time and high binding affinity, which help reduce the risk of drug resistance [271] and maintain high target occupancy even at low plasma concentrations [272]. However, selectivity remains a major challenge: high reactivity may lead to off-target modification of non-target proteins, thereby causing off-target toxicity [273]. For example, the toxicity of the UBE2N inhibitor BAY 11-7082 in multiple myeloma cells has been shown to be independent of its inhibition of the NF-κB pathway, suggesting off-target effects [274]. In contrast, research on non-covalent inhibitors targeting E2s is relatively limited. Although some molecules exhibit good activity in vitro, their pharmacokinetic properties often do not align with traditional drug design principles. For instance, non-covalent inhibitors of UBE2N such as Peptoids [275] and ML307 [276] failed to advance further in development due to poor metabolic stability or unfavorable overall pharmacokinetic profiles. Future efforts should focus on structural optimization of existing molecular scaffolds to improve key pharmacokinetic parameters such as metabolic stability while maintaining activity. Allosteric inhibitors function by binding to allosteric pockets distinct from the catalytic site, typically offering better selectivity and lower toxicity. However, allosteric sites are often concealed and difficult to identify. Despite the generally shallow active sites of E2s, which lack typical druggable features, successful cases such as the allosteric inhibitor CC0651 [277] for UBE2R1 have been reported. This molecule inhibits catalytic function by inducing conformational changes in the enzyme and distorting its active center. This strategy provides an important reference for developing highly selective inhibitors targeting other E2s. Protein-protein interaction (PPI) inhibitors theoretically offer exceptionally high specificity [278]. However, PPI interfaces are usually large, flat, and exhibit strong binding affinity, making them difficult to effectively block with small molecules. Candidate molecules in this field often face challenges such as poor solubility and high molecular weight. Currently, research in this area remains in its early stages, but preliminary breakthroughs have been made. For example, C25-140 can effectively inhibit the E3 ligase activity of TRAF6 by specifically disrupting the interaction between TRAF6 and UBE2N [279].
In addition to using small molecule inhibitors to directly target the structure of E2s, using miRNAs to inhibit E2 mRNAs may also affect the protein turnover process of cancer cells. MiRNA is an endogenous small non-coding RNA involved in the regulation of gene expression (Table 3) [280]. These molecules potently suppress the expression of their target genes, thereby inhibiting cell growth in vitro and tumor growth in vivo. Compared with plasmid DNA-based gene therapy and protein-based drug molecules, miRNA, as a natural antisense nucleotide, exhibits lower immune response and low toxicity [281]. Although miRNA drugs can target molecules that cannot be targeted by chemical drugs or antibody drugs, it is difficult to achieve ideal pharmacological effects due to the low stability of miRNA and its difficulty in delivering into cells.
miRNAs targeting E2s
| Name | Target | IMechanism of action | Cancer Type | Refs |
|---|---|---|---|---|
| miR-527 | UBE2A | Binding to 3′-UTR of UBE2A | HCC | [293] |
| miR-455-5p | UBE2B | Binding to 3′-UTR of UBE2B/UBE2V1 | OC | [294] |
| miR-17/20 | UBE2C | Binding to 3′-UTR of UBE2C | GC | [295] |
| miR661-3p | UBE2C | Binding to the 3′-UTR of UBE2C | NSCLC | [280] |
| miR-381-3p | UBE2C | Directly targeting UBE2C | PC | [296] |
| miR-140-3p | UBE2C | Directly targeting UBE2C | OS | [297] |
| miR-483-3p | UBE2C | Binding to the 3′-UTR of UBE2C | PC | [298] |
| miR-525-5p | UBE2C | Binding to the 3′-UTR of UBE2C | CRC | [221] |
| miR-379-5p | UBE2E3 | Binding to the 3′-UTR of UBE2E3 | BC | [61] |
| miR-101 | UBE2H, UBE2N | Directly targeting UBE2H/UBE2N | LUAD, BC | [299, 300] |
| miR-30a | UBE2H | Directly targeting UBE2H | LUAD | [299] |
| miR-30b | UBE2H | Directly targeting UBE2H | LUAD | [299] |
| miR-214 | UBE2I | Binding to 3′-UTR of UBE2I | GBM | [301] |
| miRNA-10a-5p | UBE2I | Binding to 3′-UTR of UBE2I | CRC | [302] |
| miR-122-3p | UBE2I | Directly targeting UBE2I | LC | [303] |
| miR-147b | UBE2N | Binding to 3′-UTR of UBE2N | HCC | [304] |
| miR-590-3p | UBE2N | Directly targeting UBE2N | CRC | [305] |
| miR-205 | UBE2N | Directly targeting UBE2N | BC | [306] |
| miR-31 | UBE2N | Directly targeting UBE2N | BC | [306] |
| miR-934 | UBE2N | Binding to 3′-UTR of UBE2N | BCa | [102] |
| miR-671-5p | UBE2R1 | Binding to 3′-UTR of UBE2R1 | OS | [104] |
| miR-1305 | UBE2T | Binding to 3′-UTR of UBE2T | HCC | [281] |
| miR-1322 | UBE2T | Binding to 3′-UTR of UBE2T | HCC | [205] |
| miR-543 | UBE2T | Directly targeting UBE2T | BC | [307] |
| miR-212-5p | UBE2T | Binding to 3′-UTR of UBE2T | HCC | [308] |
| miR-498 | UBE2T | Binding to 3′-UTR of UBE2T | MEL | [309] |
| MiR-182-5p | UBE2T | Directly targeting UBE2T | RCC | [310] |
| miR-490-5p | UBE2T | Binding to 3′-UTR of UBE2T | NSCLC | [311] |
| miR-605-5p | UBE2T | Directly targeting UBE2T | HCC | [312] |
| miR-548c-3p | UBE2T | Directly targeting UBE2T | HCC | [312] |
| miR-4689 | UBE2V1 | Directly targeting UBE2V1 | PC | [75] |
| miR-499a | UBE2V2 | Binding to 3′-UTR of UBE2V2 | PCa | [76] |
Currently, among specific components in UPP, inhibitors targeting E3s and DUBs have advanced to clinical trials for treat cancer, and inhibitors targeting E2 are still in the basic research stage [282]. Nevertheless, E2s serve as promising therapeutic targets, and their potential should not be overlooked. Therefore, further investigations need to be carried out to evaluate the feasibility and sensitivity of E2 as a potential target. E2-targeted-strategies need to be more effective, sensitive and safer as a therapeutic target for cancer.
Conclusions and Perspective
Over the past decade, our understanding of E2s within the ubiquitin-proteasome system and their roles in tumor biology has significantly deepened. While targeting E2s has emerged as a promising therapeutic strategy in oncology, their clinical translation faces critical challenges. These primarily stem from the functional complexity of E2s, their inherent "undruggability" due to suboptimal binding site characteristics, and the potential toxicity risks associated with systemic inhibition.
This review systematically elucidates that E2s, by dynamically modifying a diverse array of substrates, are integral to core processes driving tumorigenesis and progression. Notably, certain E2s, such as UBE2D3 and UBE2L3, exhibit a "dual-role" functionality in cancer. Their biological impact is highly contingent on the specific E3 ligase they cooperate with and the particular substrate they modify. For instance, UBE2D3, when paired with MDM2, catalyzes K48-linked polyubiquitination of the tumor suppressor p53, leading to its proteasomal degradation—a key mechanism enabling cancer cells to evade growth suppression and apoptosis (Figure 4C) [313]. Conversely, in concert with KLHL13, UBE2D3 mediates K63-linked polyubiquitination of TAP2 at lysine 245, thereby impairing antigen presentation pathways and anti-tumor immunity in pancreatic cancer [141]. Similarly, UBE2L3 can promote the ubiquitination and degradation of 53BP1 [314], suppressing high-fidelity homologous recombination repair and exacerbating genomic instability, while it also facilitates the degradation of GSK-3β, promoting EMT and metastasis [315]. This context-dependent duality underscores the central, yet complex, position of E2s in oncogenic signaling networks and highlights the necessity for highly precise and selective targeting strategies in future therapeutic development.
Looking ahead, overcoming the current bottlenecks in E2-targeted therapy requires a concerted effort across three dimensions. First, at the level of basic research, a deeper mechanistic dissection is imperative. Future studies must systematically delineate the functional networks, regulatory logic, and downstream signaling crosstalk of individual E2s within specific biological contexts and tumor microenvironments. This foundational knowledge is crucial for developing selective intervention strategies, such as those targeting specific E2-E3 pairs, distinct ubiquitin chain linkages, or particular substrate modification events.
Second, in drug discovery, innovative pharmacological paradigms are needed to overcome the "undruggable" nature of many E2s. The typically shallow and flat active sites of E2s pose a significant hurdle for conventional small-molecule inhibitors. Future directions should focus on: 1) developing novel chemotypes with high potency through structure-based rational design or mining of natural products; 2) embracing protein degradation technologies, such as PROTACs and molecular glues, to shift the paradigm from "functional inhibition" to "protein elimination"; and 3) exploiting covalent targeting strategies for E2s with suitable residues to enhance binding affinity and selectivity.
Third, regarding therapeutic strategy, optimized approaches are essential to balance efficacy and safety. Given the global role of E2s in maintaining proteostasis, broad inhibition may incur significant toxicity. Smarter strategies include: 1) exploring the E2-inhibitory activities of already approved drugs (e.g., ATO [316]) to expedite clinical translation, and 2) repositioning E2 inhibitors as "sensitizers" or combination partners with chemotherapy, radiotherapy, or other targeted agents. Such synergistic approaches could lower effective doses, improve therapeutic indices, and overcome resistance.
The emergence of novel therapeutic modalities is fundamentally expanding the design landscape for E2-targeting drugs. Among these, proteolysis-targeting chimeras (PROTACs [317-318]) and molecular glue technologies [319] represent a paradigm shift—from traditional "occupancy-driven inhibition" to "event-driven clearance." As bifunctional molecules, PROTACs simultaneously recruit the target protein and an E3 ligase, inducing ubiquitination and subsequent proteasomal degradation of the target. Molecular glues, in a more refined manner, induce or stabilize interactions between the target protein and an E3 ligase or other degradation machinery components, thereby "hijacking" the target into the degradation pathway. The core advantage of both strategies lies in their catalytic mechanism of action: instead of relying on prolonged target occupancy, they operate by triggering discrete degradation events. Consequently, these approaches often demonstrate high potency at low doses and hold promise for overcoming resistance caused by target overexpression or mutation.
In conclusion, E2s represent a promising frontier in cancer therapy, yet their path to clinical application is fraught with mechanistic complexity and technical hurdles. Future breakthroughs will depend on the synergistic integration of three pillars: deepening mechanistic insights, diversifying drug development technologies, and intelligent clinical strategizing. Through the convergence and collaborative innovation of structural biology, chemical biology, computational science, and clinical oncology, we can anticipate the eventual translation of E2-targeted therapies into precise and effective treatments for cancer patients.
Abbreviations
53BP1: P53-binding protein 1; ABC-DLBCL: activated B-cell-like diffuse large B-cell lymphoma; ACSL4: acyl-CoA synthetase long-chain family member 4; AKIP1: a kinase interacting protein 1; AKT: protein kinase B; AMPKα2: AMP-activated protein kinase catalytic subunit alpha 2; Apaf1: apoptotic protease-activating factor 1; APC/C: anaphase-promoting complex/cyclosome; ATP: adenosine triphosphate; Axin1: axis inhibition protein 1; BAX: BCL-2-associated X protein; BC: breast carcinoma; BCa: bladder cancer; BCL2: B-cell lymphoma 2; Bim: Bcl-2 interacting mediator of cell death; BIN1: bridging integrator 1; BRCA1: breast cancer gene 1; BRCA2: breast cancer gene 2; Caspase-3: cysteinyl aspartate-specific protease-3; Caspase-9: cysteinyl aspartate-specific protease-9; CBX6: chromobox homolog 6; CBL: Casitas B-lineage lymphoma proto-oncogene; CCNB1: cyclin B1; CCND1: cyclin D1; CDC25A: cell division cycle 25 homolog A; CDC34: cell division cycle 34; CDK1: cyclin-dependent kinase 1; CGAS: cyclic GMP-AMP synthase; CK2: casein kinase 2; CLAP1: cellular inhibitor of apoptosis protein 1; C-Maf: v-maf musculoaponeurotic fibrosarcoma oncogene homolog; CML: chronic myeloid leukemia; CNOT4: CCR4-NOT transcription complex subunit 4; CNV: copy number variation; CRC: colorectal cancer; CSF3R: colony stimulating factor 3 receptor; Cue1: coupling of ubiquitin conjugation to ER degradation protein 1; DEPTOR: DEP domain containing mTOR-interacting protein; EC: endometrial cancer; EGFR: epidermal growth factor receptor; ER: endoplasmic reticulum; ERK: extracellular signal-regulated kinase; E1: ubiquitin-activating enzyme; E2: ubiquitin-conjugating enzyme; E3: ubiquitin-protein ligase; FANCD2: fanconi anemia complementation group D2 protein; FAS: first apoptosis signal receptor; FASL: fas ligand; FL: follicular lymphoma; GBC: gallbladder carcinoma; GBM: glioblastoma; GC: gastric cancer; GCB-DLBCL: germinal center B-cell-like diffuse large B-cell lymphoma; H2AX/γH2AX: H2A histone family member X/Ser139 phosphorylated H2AX; H2B: histone H2B; H3K4me3: histone H3 lysine 4 trimethylation; H4K4: histone H4 lysine 4; HADHA: hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit alpha; HCC: hepatocellular carcinoma; HIF-1α: hypoxia inducible factor 1 subunit alpha; HMGB1: high mobility group box 1; HP1α: heterochromatin protein 1 alpha; HSP90AB1: heat shock protein 90kDa alpha, class B member 1; hTERT: human telomerase reverse transcriptase; IAP: inhibitor of apoptosis protein; ICC: intrahepatic cholangiocarcinoma; IDH1/2: isocitrate dehydrogenase 1 and 2; IFIT3: interferon-induced protein with tetratricopeptide repeats 3; IFN-I: type I interferon; IFN-γ: interferon-gamma; IGFBP3: insulin-like growth factor binding protein 3; IKK: IκB kinase; K9me3: histone H3 lysine 9 trimethylation; KAT2B: K(lysine) acetyltransferase 2B; KLHL13: Kelch-like protein 13; L3MBTL2: lethal(3) malignant brain tumor-like protein 2; LPP: LIM domain containing preferred translocation partner in lipoma; LUSC: lung squamous cell carcinoma; MDM2: mouse double minute 2; Mix1: Mix family homeobox 1; MEL: melanoma; MLL: mixed lineage leukemia; MMP-9: matrix metalloproteinase-9; Mms2: methyl methanesulfonate sensitivity protein 2; mTOR: mammalian target of rapamycin; Mule: ARF-binding protein 1; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; NOTCH: Notch receptor; NOXA: NADPH oxidase activator; Noxa: phorbol-12-myristate-13-acetate-induced protein 1; NSCLC: non-small cell lung cancer; OS: osteosarcoma; OTU: ovarian tumor domain protease; P16: cyclin-dependent kinase inhibitor 2A; P21: cyclin-dependent kinase inhibitor 1A; P27: cyclin-dependent kinase inhibitor 1B; P27Kip1: p27 kinase inhibitory protein 1; P53: tumor protein 53; PBX1: pre-B-cell leukemia homeobox 1; PC: pancreatic cancer; PCa: prostatic carcinoma; PCNA: proliferating cell nuclear antigen; PGAM1: phosphoglycerate mutase 1; PI3K: phosphatidylinositol 3-kinase; PLEKHG4: DEP domain containing mTOR-interacting protein; POT1: protection of telomeres 1; PTRF: polymerase I and transcript release factor; Puma: P53 upregulated modulator of apoptosis; RACK1: receptor for activated C kinase 1; RAD51: radiation sensitive 51; RAD52: radiation sensitive 52; RAD6: radiation sensitive 6; RECQL4: RecQ like helicase 4; RHBDF2: rhomboid 5 homolog 2; RNF19A: ring finger protein 19A; RNF213: ring finger protein 213; RPL26: ribosomal protein L26; RPL6: ribosomal protein L6; SARC: sarcoma; SCFβTRCP: skp1-Cul1-F-box protein (β-transducin repeat-containing protein) complex; SIRT1: sirtuin 1; SMAD4: suppressor of mothers against decapentaplegic homolog 4; SMAD6: SMAD family member 6; SNAT2: sodium-coupled neutral amino acid transporter 2; SNV: single nucleotide variant; SORBS3: sorbin and SH3 domain containing 3; TACE: tumor necrosis factor-α converting enzyme; TAP2: transporter associated with antigen processing 2; TGF-β: transforming growth factor-beta; TNF: tumor necrosis factor; TOP2A: DNA topoisomerase II alpha; TRAF6: TNF receptor-associated factor 6; TRF1: telomeric repeat-binding factor 1; TRF2: telomeric repeat-binding factor 2; TSC1: tuberous sclerosis complex 1; Ubl: ubiquitin-like protein 1; UPP: ubiquitin-proteasome pathway; VEGFR: vascular endothelial growth factor receptor; VEGFR2: vascular endothelial growth factor receptor 2; VEGF-A: vascular endothelial growth factor A; VHL: von Hippel-Lindau tumor suppressor; WDR76: WD repeat domain 76; β-TrCP: β-transducin repeat-containing protein.
Acknowledgements
We would like to express our sincere gratitude to all current and former members of our laboratory, as well as our collaborators, for their invaluable contributions to the work referenced in this review. Due to space limitations, we regret that we were unable to cite all relevant recent publications.
Funding
This study was supported by the General Program of the National Natural Science Foundation of China (82374092 and 82474126), Natural Science Foundation of Zhejiang Province (QKHM25H3103), "Pioneer" and "Leading Goose" R&D Program of Zhejiang (2024SDYXS0003), Program of Zhejiang Provincial TCM Sci-Tech Plan (GZY-ZJ-KJ-24064), and Zhejiang Provincial Natural Science Foundation Distinguished Young Scholar Program (LR25H280002).
Consent for publication
All authors read and approved the final manuscript.
Author contributions
Zhiyang Yao: Writing the main manuscript text and preparing figures; Ti Peng and Yong Liao: Writing the manuscript text; Hao Dong: Providing guidance on writing; Kai Miao, Jiang-Jiang Qin and Xiaoqing Guan: Providing guidance on writing, funding acquisition, and supervision.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Johnson M, Garassino MC, Mok T, Mitsudomi T. Treatment strategies and outcomes for patients with EGFR-mutant non-small cell lung cancer resistant to EGFR tyrosine kinase inhibitors: Focus on novel therapies. Lung Cancer. 2022;170:41-51
2. Minguet J, Smith KH, Bramlage P. Targeted therapies for treatment of non-small cell lung cancer-Recent advances and future perspectives. Int J Cancer. 2016;138:2549-61
3. Bizuayehu HM, Ahmed KY, Kibret GD, Dadi AF, Belachew SA, Bagade T. et al. Global Disparities of Cancer and Its Projected Burden in 2050. JAMA Netw Open. 2024;7:e2443198
4. The global challenge of cancer. Nat Cancer. 2020; 1: 1-2.
5. Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004;1695:55-72
6. Wang XK, Liao XW, Zhou X, Han CY, Chen ZJ, Yang CK. et al. Oncogene UBE2I enhances cellular invasion, migration and proliferation abilities via autophagy-related pathway resulting in poor prognosis in hepatocellular carcinoma. Am J Cancer Res. 2020;10:4178-97
7. Stewart MD, Ritterhoff T, Klevit RE, Brzovic PS. E2 enzymes: more than just middle men. Cell Res. 2016;26:423-40
8. Lim KH, Song MH, Baek KH. Decision for cell fate: deubiquitinating enzymes in cell cycle checkpoint. Cell Mol Life Sci. 2016;73:1439-55
9. Haakonsen DL, Rape M. Branching Out: Improved Signaling by Heterotypic Ubiquitin Chains. Trends Cell Biol. 2019;29:704-16
10. Vandenabeele P, Bertrand MJ. The role of the IAP E3 ubiquitin ligases in regulating pattern-recognition receptor signalling. Nat Rev Immunol. 2012;12:833-44
11. van Wijk SJ, Timmers HT. The family of ubiquitin-conjugating enzymes (E2s): deciding between life and death of proteins. Faseb j. 2010;24:981-93
12. McGinty RK, Henrici RC, Tan S. Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature. 2014;514:591-6
13. Pruneda JN, Smith FD, Daurie A, Swaney DL, Villén J, Scott JD. et al. E2~Ub conjugates regulate the kinase activity of Shigella effector OspG during pathogenesis. Embo j. 2014;33:437-49
14. Huang DT, Miller DW, Mathew R, Cassell R, Holton JM, Roussel MF. et al. A unique E1-E2 interaction required for optimal conjugation of the ubiquitin-like protein NEDD8. Nat Struct Mol Biol. 2004;11:927-35
15. Summers MK, Pan B, Mukhyala K, Jackson PK. The unique N terminus of the UbcH10 E2 enzyme controls the threshold for APC activation and enhances checkpoint regulation of the APC. Mol Cell. 2008;31:544-56
16. Zheng N, Wang P, Jeffrey PD, Pavletich NP. Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell. 2000;102:533-9
17. Dominguez C, Bonvin AM, Winkler GS, van Schaik FM, Timmers HT, Boelens R. Structural model of the UbcH5B/CNOT4 complex revealed by combining NMR, mutagenesis, and docking approaches. Structure. 2004;12:633-44
18. Kolman CJ, Toth J, Gonda DK. Identification of a portable determinant of cell cycle function within the carboxyl-terminal domain of the yeast CDC34 (UBC3) ubiquitin conjugating (E2) enzyme. Embo j. 1992;11:3081-90
19. Spratt DE, Shaw GS. Association of the disordered C-terminus of CDC34 with a catalytically bound ubiquitin. J Mol Biol. 2011;407:425-38
20. Sadowski M, Mawson A, Baker R, Sarcevic B. Cdc34 C-terminal tail phosphorylation regulates Skp1/cullin/F-box (SCF)-mediated ubiquitination and cell cycle progression. Biochem J. 2007;405:569-81
21. Ravid T, Hochstrasser M. Autoregulation of an E2 enzyme by ubiquitin-chain assembly on its catalytic residue. Nat Cell Biol. 2007;9:422-7
22. Block K, Boyer TG, Yew PR. Phosphorylation of the human ubiquitin-conjugating enzyme, CDC34, by casein kinase 2. J Biol Chem. 2001;276:41049-58
23. Ceccaldi R, Sarangi P, D'Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol. 2016;17:337-49
24. Ikeda F, Deribe YL, Skånland SS, Stieglitz B, Grabbe C, Franz-Wachtel M. et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature. 2011;471:637-41
25. Li X, Elmira E, Rohondia S, Wang J, Liu J, Dou QP. A patent review of the ubiquitin ligase system: 2015-2018. Expert Opin Ther Pat. 2018;28:919-37
26. Zhang G. The mutation rate as an evolving trait. Nat Rev Genet. 2023;24:3
27. Bergeron LA, Besenbacher S, Turner T, Versoza CJ, Wang RJ, Price AL. et al. The Mutationathon highlights the importance of reaching standardization in estimates of pedigree-based germline mutation rates. Elife. 2022 11
28. Casolino R, Beer PA, Chakravarty D, Davis MB, Malapelle U, Mazzarella L. et al. Interpreting and integrating genomic tests results in clinical cancer care: Overview and practical guidance. CA Cancer J Clin. 2024;74:264-85
29. Cosman D. The hematopoietin receptor superfamily. Cytokine. 1993;5:95-106
30. Magistroni V, Mauri M, D'Aliberti D, Mezzatesta C, Crespiatico I, Nava M. et al. De novo UBE2A mutations are recurrently acquired during chronic myeloid leukemia progression and interfere with myeloid differentiation pathways. Haematologica. 2019;104:1789-97
31. Ma G, Gao Y, Jing X, He C, Liu H, Wu X. et al. Targeted sequencing reveals the relationship between mutations and patients' clinical indicators, blood cell counts and early progression in diffuse large-B cell lymphoma. Leuk Lymphoma. 2023;64:140-50
32. de Miranda NF, Georgiou K, Chen L, Wu C, Gao Z, Zaravinos A. et al. Exome sequencing reveals novel mutation targets in diffuse large B-cell lymphomas derived from Chinese patients. Blood. 2014;124:2544-53
33. González-Rincón J, Méndez M, Gómez S, García JF, Martín P, Bellas C. et al. Unraveling transformation of follicular lymphoma to diffuse large B-cell lymphoma. PLoS One. 2019;14:e0212813
34. Wang C, Bai R, Liu Y, Wang K, Wang Y, Yang J. et al. Multi-region sequencing depicts intratumor heterogeneity and clonal evolution in cervical cancer. Med Oncol. 2023;40:78
35. Hao Z, Zhang H, Cowell J. Ubiquitin-conjugating enzyme UBE2C: molecular biology, role in tumorigenesis, and potential as a biomarker. Tumour Biol. 2012;33:723-30
36. Ma S, Chen Q, Li X, Fu J, Zhao L. UBE2C serves as a prognosis biomarker of uterine corpus endometrial carcinoma via promoting tumor migration and invasion. Sci Rep. 2023;13:16899
37. Menezes J, Acquadro F, Wiseman M, Gómez-López G, Salgado RN, Talavera-Casañas JG. et al. Exome sequencing reveals novel and recurrent mutations with clinical impact in blastic plasmacytoid dendritic cell neoplasm. Leukemia. 2014;28:823-9
38. Everett RD, Boutell C, Hale BG. Interplay between viruses and host sumoylation pathways. Nat Rev Microbiol. 2013;11:400-11
39. Seeler JS, Dejean A. SUMO and the robustness of cancer. Nat Rev Cancer. 2017;17:184-97
40. Weile J, Sun S, Cote AG, Knapp J, Verby M, Mellor JC. et al. A framework for exhaustively mapping functional missense variants. Mol Syst Biol. 2017;13:957
41. Zhang J, Kinch LN, Cong Q, Weile J, Sun S, Cote AG. et al. Assessing predictions of fitness effects of missense mutations in SUMO-conjugating enzyme UBE2I. Hum Mutat. 2017;38:1051-63
42. Hu Q, Li X, Wang P, Xie Y. Pan-cancer analysis unveils the role and mechanisms of neddylation modifications in tumorigenesis. Med Oncol. 2025;42:119
43. Ullah K, Zubia E, Narayan M, Yang J, Xu G. Diverse roles of the E2/E3 hybrid enzyme UBE2O in the regulation of protein ubiquitination, cellular functions, and disease onset. Febs j. 2019;286:2018-34
44. Vila IK, Yao Y, Kim G, Xia W, Kim H, Kim SJ. et al. A UBE2O-AMPKα2 Axis that Promotes Tumor Initiation and Progression Offers Opportunities for Therapy. Cancer Cell. 2017;31:208-24
45. Zhang RY, Liu ZK, Wei D, Yong YL, Lin P, Li H. et al. UBE2S interacting with TRIM28 in the nucleus accelerates cell cycle by ubiquitination of p27 to promote hepatocellular carcinoma development. Signal Transduct Target Ther. 2021;6:64
46. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
47. Ueki T, Park JH, Nishidate T, Kijima K, Hirata K, Nakamura Y. et al. Ubiquitination and downregulation of BRCA1 by ubiquitin-conjugating enzyme E2T overexpression in human breast cancer cells. Cancer Res. 2009;69:8752-60
48. Virts EL, Jankowska A, Mackay C, Glaas MF, Wiek C, Kelich SL. et al. AluY-mediated germline deletion, duplication and somatic stem cell reversion in UBE2T defines a new subtype of Fanconi anemia. Hum Mol Genet. 2015;24:5093-108
49. Yuan Y, Xiao WW, Xie WH, Li RZ, Gao YH. Prognostic value of ubiquitin E2 UBE2W and its correlation with tumor-infiltrating immune cells in breast cancer. BMC Cancer. 2021;21:479
50. Yang H, Ye D, Guan KL, Xiong Y. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res. 2012;18:5562-71
51. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM. et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739-44
52. Ward PS, Cross JR, Lu C, Weigert O, Abel-Wahab O, Levine RL. et al. Identification of additional IDH mutations associated with oncometabolite R(-)-2-hydroxyglutarate production. Oncogene. 2012;31:2491-8
53. Ramatenki V, Potlapally SR, Dumpati RK, Vadija R, Vuruputuri U. Homology modeling and virtual screening of ubiquitin conjugation enzyme E2A for designing a novel selective antagonist against cancer. J Recept Signal Transduct Res. 2015;35:536-49
54. Kao WC, Hsu SH, Lin CL, Lin CY, Chen SW, Chen YX. et al. Role of high ubiquitin-conjugating enzyme E2 expression as a prognostic factor in nasopharyngeal carcinoma. Oncol Lett. 2022;23:194
55. Zhang S, You X, Zheng Y, Shen Y, Xiong X, Sun Y. The UBE2C/CDH1/DEPTOR axis is an oncogene and tumor suppressor cascade in lung cancer cells. J Clin Invest. 2023 133
56. Li L, Li X, Wang W, Gao T, Shi Z. UBE2C is involved in the functions of ECRG4 on esophageal squamous cell carcinoma. Biomed Pharmacother. 2018;98:201-6
57. Pan Z, Bao J, Zhang L, Wei S. UBE2D3 Activates SHP-2 Ubiquitination to Promote Glycolysis and Proliferation of Glioma via Regulating STAT3 Signaling Pathway. Front Oncol. 2021;11:674286
58. Lei K, Zhao Y, Li S, Liu J, Chen W, Zhou C. et al. Integrative spatial and single-cell transcriptomics elucidate programmed cell death-driven tumor microenvironment dynamics in hepatocellular carcinoma. Front Immunol. 2025;16:1589563
59. Wheaton K, Sarkari F, Stanly Johns B, Davarinejad H, Egorova O, Kaustov L. et al. UbE2E1/UBCH6 Is a Critical in vivo E2 for the PRC1-catalyzed Ubiquitination of H2A at Lys-119. J Biol Chem. 2017;292:2893-902
60. Mizukami S, Watanabe Y, Saegusa Y, Nakajima K, Ito Y, Masubuchi Y. et al. Downregulation of UBE2E2 in rat liver cells after hepatocarcinogen treatment facilitates cell proliferation and slowing down of DNA damage response in GST-P-expressing preneoplastic lesions. Toxicol Appl Pharmacol. 2017;334:207-16
61. Schroder AK, Loy CJ, Aiala F, Rafique J, Ghosh A, Yoo LI. miR-379-5p affects breast cancer cell behavior by targeting UBE2E3 ubiquitin conjugating enzyme. PLoS One. 2024;19:e0310315
62. Zhang F, Xiong X, Li Z, Wang H, Wang W, Zhao Y. et al. RHEB neddylation by the UBE2F-SAG axis enhances mTORC1 activity and aggravates liver tumorigenesis. Embo j. 2025;44:1185-219
63. Dong M, Pang X, Xu Y, Wen F, Zhang Y. Ubiquitin-conjugating enzyme 9 promotes epithelial ovarian cancer cell proliferation in vitro. Int J Mol Sci. 2013;14:11061-71
64. Tian Y, Dong R, Guan Y, Wang Y, Zhao W, Zhang J. et al. UBE2J1 is identified as a novel plasma cell-related gene involved in the prognosis of high-grade serous ovarian cancer. J Transl Med. 2025;23:129
65. Wang T, Jin C, Yang P, Chen Z, Ji J, Sun Q. et al. UBE2J1 inhibits colorectal cancer progression by promoting ubiquitination and degradation of RPS3. Oncogene. 2023;42:651-64
66. Khalil MI, Yang C, Vu L, Chadha S, Nabors H, James CD. et al. The membrane-associated ubiquitin ligase MARCHF8 stabilizes the human papillomavirus oncoprotein E7 by degrading CUL1 and UBE2L3 in head and neck cancer. J Virol. 2024;98:e0172623
67. Zhao B, Gao C, Shi D, Mao J, Zhao J, Guo L. et al. Knockdown of Nedd8-conjugating enzyme UBE2M suppresses the proliferation and induces the apoptosis of intrahepatic cholangiocarcinoma cells. Oncol Rep. 2019;42:2670-9
68. Liu X, Ma F, Liu C, Zhu K, Li W, Xu Y. et al. UBE2O promotes the proliferation, EMT and stemness properties of breast cancer cells through the UBE2O/AMPKα2/mTORC1-MYC positive feedback loop. Cell Death Dis. 2020;11:10
69. Tian C, Chen Z, Wang L, Si J, Kang J, Li Y. et al. Over expression of ubiquitin-conjugating enzyme E2O in bone marrow mesenchymal stromal cells partially attenuates acute myeloid leukaemia progression. Br J Haematol. 2023;200:476-88
70. Chang R, Wei L, Lu Y, Cui X, Lu C, Liu L. et al. Upregulated expression of ubiquitin-conjugating enzyme E2Q1 (UBE2Q1) is associated with enhanced cell proliferation and poor prognosis in human hapatocellular carcinoma. J Mol Histol. 2015;46:45-56
71. Maeda H, Miyajima N, Kano S, Tsukiyama T, Okumura F, Fukuda S. et al. Ubiquitin-conjugating enzyme UBE2Q2 suppresses cell proliferation and is down-regulated in recurrent head and neck cancer. Mol Cancer Res. 2009;7:1553-62
72. Liu X, Zhang Y, Hu Z, Li Q, Yang L, Xu G. The Catalytically Inactive Mutation of the Ubiquitin-Conjugating Enzyme CDC34 Affects its Stability and Cell Proliferation. Protein J. 2018;37:132-43
73. Liu F, Zhu C, Gao P, Zheng S, Li C. Ubiquitin-conjugating enzyme E2T regulates cell proliferation and migration in cholangiocarcinoma. Anticancer Drugs. 2020;31:836-46
74. Deng Y, Chen X, Chen X, Huang C, Zhang Z, Xu Z. et al. UBE2T promotes stage I lung adenocarcinoma progression through PBX1 ubiquitination and PBX1/RORA regulation. BMC Cancer. 2024;24:1158
75. Zhang R, Wang X, Ying X, Huang Y, Zhai S, Shi M. et al. Hypoxia-induced long non-coding RNA LINC00460 promotes p53 mediated proliferation and metastasis of pancreatic cancer by regulating the miR-4689/UBE2V1 axis and sequestering USP10. Int J Med Sci. 2023;20:1339-57
76. Chen Y, Sun F, Zhang L, Zhou J, Hou J. miR-499a inhibits the proliferation and apoptosis of prostate cancer via targeting UBE2V2. World J Surg Oncol. 2021;19:250
77. Yang Z, Wu G, Zhao J, Shi G, Zhou J, Zhou X. UBE2V2 promotes metastasis by regulating EMT and predicts a poor prognosis in lung adenocarcinoma. Cancer Med. 2023;12:19850-65
78. Shi X, Wang B, Chen X, Zheng Y, Ding Y, Wang C. Upregulation of ubiquitin-conjugating enzyme E2Z is associated with human hepatocellular carcinoma. Biochem Biophys Res Commun. 2020;523:25-32
79. Lv Z, Wang M, Hou H, Tang G, Xu H, Wang X. et al. FOXM1-regulated ZIC2 promotes the malignant phenotype of renal clear cell carcinoma by activating UBE2C/mTOR signaling pathway. Int J Biol Sci. 2023;19:3293-306
80. Zhang Z, Liu P, Wang J, Gong T, Zhang F, Ma J. et al. Ubiquitin-conjugating enzyme E2C regulates apoptosis-dependent tumor progression of non-small cell lung cancer via ERK pathway. Med Oncol. 2015;32:149
81. Critchley WR, Smith GA, Zachary IC, Harrison MA, Ponnambalam S. The E2 ubiquitin-conjugating enzymes UBE2D1 and UBE2D2 regulate VEGFR2 dynamics and endothelial function. J Cell Sci. 2023 136
82. Obacz J, Archambeau J, Lafont E, Nivet M, Martin S, Aubry M. et al. IRE1 endoribonuclease signaling promotes myeloid cell infiltration in glioblastoma. Neuro Oncol. 2024;26:858-71
83. Sun L, Amraei R, Rahimi N. NEDD4 regulates ubiquitination and stability of the cell adhesion molecule IGPR-1 via lysosomal pathway. J Biomed Sci. 2021;28:35
84. Chen X, Zhang S, Liu C, Li G, Lu S, Wang Y. et al. UBE2O Promotes Progression and Epithelial-Mesenchymal Transition in Head and Neck Squamous Cell Carcinoma. Onco Targets Ther. 2020;13:6191-202
85. Gao X, Wang W, Yang H, Wu L, He Z, Zhou S. et al. UBE2D3 gene overexpression increases radiosensitivity of EC109 esophageal cancer cells in vitro and in vivo. Oncotarget. 2016;7:32543-53
86. Yalçin Z, Lam SY, Peuscher MH, van der Torre J, Zhu S, Iyengar PV. et al. UBE2D3 facilitates NHEJ by orchestrating ATM signalling through multi-level control of RNF168. Nat Commun. 2024;15:5032
87. Kim S, Park SH, Kang N, Ra JS, Myung K, Lee KY. Polyubiquitinated PCNA triggers SLX4-mediated break-induced replication in alternative lengthening of telomeres (ALT) cancer cells. Nucleic Acids Res. 2024;52:11785-805
88. Margalef P, Kotsantis P, Borel V, Bellelli R, Panier S, Boulton SJ. Stabilization of Reversed Replication Forks by Telomerase Drives Telomere Catastrophe. Cell. 2018;172:439-53.e14
89. Zhou F, Sun Y, Chen X, Wu D, Tao Z, Wu L. et al. UBE2T-Mediated HP1α Ubiquitination Enhances Nucleolar Function and Promotes the Progression of IDH1/TP53-Mutant Glioma. Clin Cancer Res. 2025;31:3970-83
90. Perrotta I, Bruno L, Maltese L, Russo E, Donato A, Donato G. Immunohistochemical analysis of the ubiquitin-conjugating enzyme UbcH10 in lung cancer: a useful tool for diagnosis and therapy. J Histochem Cytochem. 2012;60:359-65
91. Ma Z, Liu Y, Hao Z, Hua X, Li W. DNA hypermethylation of aurora kinase A in hepatitis C virus-positive hepatocellular carcinoma. Mol Med Rep. 2019;20:2519-32
92. Huang X, Tao Y, Gao J, Zhou X, Tang S, Deng C. et al. UBC9 coordinates inflammation affecting development of bladder cancer. Sci Rep. 2020;10:20670
93. Mayca Pozo F, Geng X, Tamagno I, Jackson MW, Heimsath EG, Hammer JA. et al. MYO10 drives genomic instability and inflammation in cancer. Sci Adv. 2021;7:eabg6908
94. Wang G, Hua R, Chen X, He X, Dingming Y, Chen H. et al. MX1 and UBE2L6 are potential metaflammation gene targets in both diabetes and atherosclerosis. PeerJ. 2024;12:e16975
95. Xu M, Tan J, Zhu L, Ge C, Dong W, Dai X. et al. The deubiquitinating enzyme 13 retards non-alcoholic steatohepatitis via blocking inactive rhomboid protein 2-dependent pathway. Acta Pharm Sin B. 2023;13:1071-92
96. Wang Y, Ma Q, Li H, Huang W, You J, Liu D. UBE2D1 promotes glioblastoma proliferation by modulating p21 ubiquitination. Mol Carcinog. 2024;63:1967-79
97. Plafker KS, Zyla K, Berry W, Plafker SM. Loss of the ubiquitin conjugating enzyme UBE2E3 induces cellular senescence. Redox Biol. 2018;17:411-22
98. Chang Y, Chen Q, Li H, Xu J, Tan M, Xiong X. et al. The UBE2F-CRL5(ASB11)-DIRAS2 axis is an oncogene and tumor suppressor cascade in pancreatic cancer cells. Dev Cell. 2024;59:1317-32.e5
99. Huang J, Tan X, Liu Y, Jiang K, Luo J. Knockdown of UBE2I inhibits tumorigenesis and enhances chemosensitivity of cholangiocarcinoma via modulating p27kip1 nuclear export. Mol Carcinog. 2023;62:700-15
100. Ma X, Zhao J, Yang F, Liu H, Qi W. Ubiquitin conjugating enzyme E2 L3 promoted tumor growth of NSCLC through accelerating p27kip1 ubiquitination and degradation. Oncotarget. 2017;8:84193-203
101. Zhou W, Xu J, Tan M, Li H, Li H, Wei W. et al. UBE2M Is a Stress-Inducible Dual E2 for Neddylation and Ubiquitylation that Promotes Targeted Degradation of UBE2F. Mol Cell. 2018;70:1008-24.e6
102. Yan H, Ren S, Lin Q, Yu Y, Chen C, Hua X. et al. Inhibition of UBE2N-dependent CDK6 protein degradation by miR-934 promotes human bladder cancer cell growth. Faseb j. 2019;33:12112-23
103. Fahmidehkar MA, Shafiee SM, Eftekhar E, Mahbudi L, Seghatoleslam A. Induction of cell proliferation, clonogenicity and cell accumulation in S phase as a consequence of human UBE2Q1 overexpression. Oncol Lett. 2016;12:2169-74
104. Xin C, Lu S, Li Y, Zhang Y, Tian J, Zhang S. et al. miR-671-5p Inhibits Tumor Proliferation by Blocking Cell Cycle in Osteosarcoma. DNA Cell Biol. 2019;38:996-1004
105. Zhang M, Liu Y, Yin Y, Sun Z, Wang Y, Zhang Z. et al. UBE2S promotes the development of ovarian cancer by promoting PI3K/AKT/mTOR signaling pathway to regulate cell cycle and apoptosis. Mol Med. 2022;28:62
106. Guo J, Wang M, Wang JP, Wu CX. Ubiquitin-conjugating enzyme E2T knockdown suppresses hepatocellular tumorigenesis via inducing cell cycle arrest and apoptosis. World J Gastroenterol. 2019;25:6386-403
107. Rosner K, Mehregan DR, Kirou E, Abrams J, Kim S, Campbell M. et al. Melanoma Development and Progression Are Associated with Rad6 Upregulation and β -Catenin Relocation to the Cell Membrane. J Skin Cancer. 2014;2014:439205
108. Ding H, Xu JC, Ding ZG, Wu LF, Liu YB, Zhang YF. et al. Identification and validation of UBE2B as a prognostic biomarker promoting the development of esophageal carcinomas. Front Immunol. 2024;15:1295305
109. Tang XK, Wang KJ, Tang YK, Chen L. Effects of ubiquitin-conjugating enzyme 2C on invasion, proliferation and cell cycling of lung cancer cells. Asian Pac J Cancer Prev. 2014;15:3005-9
110. Li W, Chen C, Zheng H, Lin Y, An M, Liu D. et al. UBE2C-induced crosstalk between mono- and polyubiquitination of SNAT2 promotes lymphatic metastasis in bladder cancer. J Clin Invest. 2024 134
111. Zhi J, Sun J, Wang Z, Ding W. Support vector machine classifier for prediction of the metastasis of colorectal cancer. Int J Mol Med. 2018;41:1419-26
112. Hong X, Ma N, Li D, Zhang M, Dong W, Huang J. et al. UBE2E2 enhances Snail-mediated epithelial-mesenchymal transition and Nrf2-mediated antioxidant activity in ovarian cancer. Cell Death Dis. 2023;14:100
113. Nikolic I, Cursons J, Shields B, Chappaz S, Sudholz H, Meng X. et al. Enhancing anti-tumor immunity of natural killer cells through targeting IL-15R signaling. Cancer Cell. 2025;43:2034-50.e11
114. Wang Q, Li S, Xu Y, Chen Y, Xu C, He Q. et al. UBC9 overexpression promotes proliferation and metastasis in gastric cancer via ATF2. World J Surg Oncol. 2025;23:270
115. Lan SY, Ding Y, Wang C, Fang J, Ren C, Liu JL. et al. High Level of Ubiquitin Conjugate Enzyme E2O Indicates Poor Prognosis of Patients with Hepatocellular Carcinoma. Curr Med Sci. 2023;43:93-103
116. Wu M, Li X, Huang W, Chen Y, Wang B, Liu X. Ubiquitin-conjugating enzyme E2T(UBE2T) promotes colorectal cancer progression by facilitating ubiquitination and degradation of p53. Clin Res Hepatol Gastroenterol. 2021;45:101493
117. Chen S, Wang DL, Liu Y, Zhao L, Sun FL. RAD6 regulates the dosage of p53 by a combination of transcriptional and posttranscriptional mechanisms. Mol Cell Biol. 2012;32:576-87
118. Huang S, Li X. UBE2C promotes LUAD progression by ubiquitin-dependent degradation of p53 to inactivate the p53/p21 signaling pathway. Discov Oncol. 2024;15:589
119. Kariri Y, Toss MS, Alsaleem M, Elsharawy KA, Joseph C, Mongan NP. et al. Ubiquitin-conjugating enzyme 2C (UBE2C) is a poor prognostic biomarker in invasive breast cancer. Breast Cancer Res Treat. 2022;192:529-39
120. Yang Y, Xu J, Zhao Q, Zhang R, Wang Y, Shang C. et al. UBE2D2 promotes gastric cancer progression by inhibiting ferroptosis through autophagy-dependent stabilization of CST1. Int J Biol Macromol. 2025;327:147333
121. Ray P, Nancarrow DJ, Ferrer-Torres D, Wang Z, San Martinho M, Hinton T. et al. UBCH5 Family Members Differentially Impact Stabilization of Mutant p53 via RNF128 Iso1 During Barrett's Progression to Esophageal Adenocarcinoma. Cell Mol Gastroenterol Hepatol. 2022;13:129-49
122. Zhou L, Zhu J, Chen W, Jiang Y, Hu T, Wang Y. et al. Induction of NEDD8-conjugating enzyme E2 UBE2F by platinum protects lung cancer cells from apoptosis and confers to platinum-insensitivity. Cell Death Dis. 2020;11:975
123. Flores-Pérez A, Elizondo G. Apoptosis induction and inhibition of HeLa cell proliferation by alpha-naphthoflavone and resveratrol are aryl hydrocarbon receptor-independent. Chem Biol Interact. 2018;281:98-105
124. Tao NN, Zhang ZZ, Ren JH, Zhang J, Zhou YJ, Wai Wong VK. et al. Overexpression of ubiquitin-conjugating enzyme E2 L3 in hepatocellular carcinoma potentiates apoptosis evasion by inhibiting the GSK3β/p65 pathway. Cancer Lett. 2020;481:1-14
125. Zhou X, Wei J, Chen F, Xiao X, Huang T, He Q. et al. Epigenetic downregulation of the ISG15-conjugating enzyme UbcH8 impairs lipolysis and correlates with poor prognosis in nasopharyngeal carcinoma. Oncotarget. 2015;6:41077-91
126. Monda JK, Cheeseman IM. Dynamic regulation of dynein localization revealed by small molecule inhibitors of ubiquitination enzymes. Open Biol. 2018 8
127. Pan YH, Yang M, Liu LP, Wu DC, Li MY, Su SG. UBE2S enhances the ubiquitination of p53 and exerts oncogenic activities in hepatocellular carcinoma. Biochem Biophys Res Commun. 2018;503:895-902
128. Cai F, Xu H, Song S, Wang G, Zhang Y, Qian J. et al. Knockdown of Ubiquitin-Conjugating Enzyme E2 T Abolishes the Progression of Head and Neck Squamous Cell Carcinoma by Inhibiting NF-Κb Signaling and inducing Ferroptosis. Curr Protein Pept Sci. 2024;25:577-85
129. Saadat N, Liu F, Haynes B, Nangia-Makker P, Bao X, Li J. et al. Nano-delivery of RAD6/Translesion Synthesis Inhibitor SMI#9 for Triple-negative Breast Cancer Therapy. Mol Cancer Ther. 2018;17:2586-97
130. Cao JZ, Nie G, Hu H, Zhang X, Ni CM, Huang ZP. et al. UBE2C promotes the progression of pancreatic cancer and glycolytic activity via EGFR stabilization-mediated PI3K-Akt pathway activation. J Gastrointest Oncol. 2022;13:1444-53
131. Yang YF, Chang YC, Tsai KW, Hung MH, Kang BH. UBE2C triggers HIF-1α-glycolytic flux in head and neck squamous cell carcinoma. J Cell Mol Med. 2022;26:3716-25
132. Jin Z, Zhao X, Cui L, Xu X, Zhao Y, Younai F. et al. UBE2C promotes the progression of head and neck squamous cell carcinoma. Biochem Biophys Res Commun. 2020;523:389-97
133. Meng Z, Bian X, Ma L, Zhang G, Ma Q, Xu Q. et al. UBC9 stabilizes PFKFB3 to promote aerobic glycolysis and proliferation of glioblastoma cells. Int J Biochem Cell Biol. 2023;165:106491
134. Yang B, Chen W, Tao T, Zhang J, Kong D, Hao J. et al. UBE2N promotes cell viability and glycolysis by promoting Axin1 ubiquitination in prostate cancer cells. Biol Direct. 2024;19:35
135. Lin J, Wang X, Zhai S, Shi M, Peng C, Deng X. et al. Hypoxia-induced exosomal circPDK1 promotes pancreatic cancer glycolysis via c-myc activation by modulating miR-628-3p/BPTF axis and degrading BIN1. J Hematol Oncol. 2022;15:128
136. Meng F, Luo X, Li C, Wang G. LncRNA LINC00525 activates HIF-1α through miR-338-3p / UBE2Q1 / β-catenin axis to regulate the Warburg effect in colorectal cancer. Bioengineered. 2022;13:2554-67
137. Wu X, Chen Y, He W, Yao Y, Liu Y, Xia P. et al. UBE2Q2 promotes tumor progression and glycolysis of hepatocellular carcinoma through NF-κB/HIF1α signal pathway. Cell Oncol (Dordr). 2025;48:637-54
138. Zhang R, Li C, Zhang S, Kong L, Liu Z, Guo Y. et al. UBE2S promotes glycolysis in hepatocellular carcinoma by enhancing E3 enzyme-independent polyubiquitination of VHL. Clin Mol Hepatol. 2024;30:771-92
139. Qiao L, Dong C, Ma B. UBE2T promotes proliferation, invasion and glycolysis of breast cancer cells by regualting the PI3K/AKT signaling pathway. J Recept Signal Transduct Res. 2022;42:151-9
140. Xu Y, Lan F, Bi Q, Li X, Wang Z, Li Y. et al. Comprehensive analysis of the prognosis and tumor immune microenvironment of ubiquitin-conjugating enzyme transport-related gene UBE2C in hepatocellular carcinoma. Discov Oncol. 2025;16:884
141. Wang S, Yang W, Peng T, Zhu C, Dong J, Wang Y. et al. IFN-γ-driven UBE2D3 upregulation impairs antigen presentation pathways and anti-tumor immunity in pancreatic cancer. Nat Commun. 2025;16:10733
142. Xiao J, Sun F, Wang YN, Liu B, Zhou P, Wang FX. et al. UBC9 deficiency enhances immunostimulatory macrophage activation and subsequent antitumor T cell response in prostate cancer. J Clin Invest. 2023 133
143. Wu D, Li H, Liu M, Qin J, Sun Y. The Ube2m-Rbx1 neddylation-Cullin-RING-Ligase proteins are essential for the maintenance of Regulatory T cell fitness. Nat Commun. 2022;13:3021
144. Sun S, Ni J, Liu J, Tan J, Jin R, Li H. et al. Ubiquitin-Conjugating Enzyme Ubc13 in Macrophages Suppresses Lung Tumor Progression Through Inhibiting PD-L1 Expression. Eur J Immunol. 2025;55:e202451118
145. Jie XL, Wei JC, Wang D, Zhang XW, Lv MY, Lin YF. et al. CDC34 suppresses macrophage phagocytic activity and predicts poor response to immune checkpoint inhibitor in cancers. Cancer Lett. 2025;628:217822
146. Bao H, Luo Y, Ding G, Fu Z. A Pan-Cancer Analysis of UBE2S in Tumorigenesis, Prognosis, Pathway, Immune Infiltration and Evasion, and Therapy Response from an Immune-Oncology Perspective. J Oncol. 2022;2022:3982539
147. Xu F, Xiong N, Yuan Y, Liu J. Prognostic Value of UBE2T and Its Correlation with Immune Infiltrates in Lung Adenocarcinoma. J Oncol. 2022;2022:5244820
148. Hua ZD, Liu XB, Sheng JH, Li C, Li P, Cai XQ. et al. UBE2V2 Positively Correlates With PD-L1 Expression and Confers Poor Patient Survival in Lung Adenocarcinoma. Appl Immunohistochem Mol Morphol. 2021;29:585-91
149. Somasagara RR, Spencer SM, Tripathi K, Clark DW, Mani C, Madeira da Silva L. et al. RAD6 promotes DNA repair and stem cell signaling in ovarian cancer and is a promising therapeutic target to prevent and treat acquired chemoresistance. Oncogene. 2017;36:6680-90
150. Kim JH, Lee KW, Yang HJ, Park JJ, Lee CH, Jeon YS. et al. The correlation between the expression of ubiquitin-conjugating enzyme 2C and prostate cancer prognosis. J Cancer Res Clin Oncol. 2023;149:6351-60
151. Bajaj S, Alam SK, Roy KS, Datta A, Nath S, Roychoudhury S. E2 Ubiquitin-conjugating Enzyme, UBE2C Gene, Is Reciprocally Regulated by Wild-type and Gain-of-Function Mutant p53. J Biol Chem. 2016;291:14231-47
152. van Ree JH, Jeganathan KB, Malureanu L, van Deursen JM. Overexpression of the E2 ubiquitin-conjugating enzyme UbcH10 causes chromosome missegregation and tumor formation. J Cell Biol. 2010;188:83-100
153. Li F, Dai Y, Tang C, Peng L, Huang H, Chen Y. et al. Elevated UBC9 expression and its oncogenic role in colorectal cancer progression and chemoresistance. Sci Rep. 2025;15:9123
154. Ouyang J, Garner E, Hallet A, Nguyen HD, Rickman KA, Gill G. et al. Noncovalent interactions with SUMO and ubiquitin orchestrate distinct functions of the SLX4 complex in genome maintenance. Mol Cell. 2015;57:108-22
155. Nakada S, Tai I, Panier S, Al-Hakim A, Iemura S, Juang YC. et al. Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature. 2010;466:941-6
156. Bonanno L, Costa C, Majem M, Sanchez JJ, Rodriguez I, Gimenez-Capitan A. et al. Combinatory effect of BRCA1 and HERC2 expression on outcome in advanced non-small-cell lung cancer. BMC Cancer. 2016;16:312
157. Paul A, Wang B. RNF8- and Ube2S-Dependent Ubiquitin Lysine 11-Linkage Modification in Response to DNA Damage. Mol Cell. 2017;66:458-72.e5
158. Sun J, Zhu Z, Li W, Shen M, Cao C, Sun Q. et al. UBE2T-regulated H2AX monoubiquitination induces hepatocellular carcinoma radioresistance by facilitating CHK1 activation. J Exp Clin Cancer Res. 2020;39:222
159. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
160. Wang L, Chen YJ, Hou J, Wang YY, Tang WQ, Shen XZ. et al. Expression and clinical significance of BIRC6 in human epithelial ovarian cancer. Tumour Biol. 2014;35:4891-6
161. Nakayama Y, Yamaguchi N. Role of cyclin B1 levels in DNA damage and DNA damage-induced senescence. Int Rev Cell Mol Biol. 2013;305:303-37
162. Monga SP. Role of Wnt/β-catenin signaling in liver metabolism and cancer. Int J Biochem Cell Biol. 2011;43:1021-9
163. Deng Y, Li Y, Wu T, Chen X, Li X, Cai K. et al. RAD6 Positively Affects Tumorigenesis of Esophageal Squamous Cell Carcinoma by Regulating Histone Ubiquitination of CCNB1. Biol Proced Online. 2022;24:4
164. Liu G, Zhao J, Pan B, Ma G, Liu L. UBE2C overexpression in melanoma and its essential role in G2/M transition. J Cancer. 2019;10:2176-84
165. Liu LL, Zhu JM, Yu XN, Zhu HR, Shi X, Bilegsaikhan E. et al. UBE2T promotes proliferation via G2/M checkpoint in hepatocellular carcinoma. Cancer Manag Res. 2019;11:8359-70
166. Ferrer JL, Dupuy J, Borel F, Jacquamet L, Noel JP, Dulic V. Structural basis for the modulation of CDK-dependent/independent activity of cyclin D1. Cell Cycle. 2006;5:2760-8
167. Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323-30
168. Cai F, Chen P, Chen L, Biskup E, Liu Y, Chen PC. et al. Human RAD6 promotes G1-S transition and cell proliferation through upregulation of cyclin D1 expression. PLoS One. 2014;9:e113727
169. Yang H, Wu L, Ke S, Wang W, Yang L, Gao X. et al. Downregulation of Ubiquitin-conjugating Enzyme UBE2D3 Promotes Telomere Maintenance and Radioresistance of Eca-109 Human Esophageal Carcinoma Cells. J Cancer. 2016;7:1152-62
170. Zhang GC, Yu XN, Sun JL, Xiong J, Yang YJ, Jiang XM. et al. UBE2M promotes cell proliferation via the β-catenin/cyclin D1 signaling in hepatocellular carcinoma. Aging (Albany NY). 2020;12:2373-92
171. Zhou C, Bi F, Yuan J, Yang F, Sun S. Gain of UBE2D1 facilitates hepatocellular carcinoma progression and is associated with DNA damage caused by continuous IL-6. J Exp Clin Cancer Res. 2018;37:290
172. Li L, Kang J, Zhang W, Cai L, Wang S, Liang Y. et al. Validation of NEDD8-conjugating enzyme UBC12 as a new therapeutic target in lung cancer. EBioMedicine. 2019;45:81-91
173. Matsui WH. Cancer stem cell signaling pathways. Medicine (Baltimore). 2016;95:S8-s19
174. Nusse R, Clevers H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell. 2017;169:985-99
175. Li Z, Wang Y, Li Y, Yin W, Mo L, Qian X. et al. Ube2s stabilizes β-Catenin through K11-linked polyubiquitination to promote mesendoderm specification and colorectal cancer development. Cell Death Dis. 2018;9:456
176. Liu J, Xiao Q, Xiao J, Niu C, Li Y, Zhang X. et al. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct Target Ther. 2022;7:3
177. Sarma A, Gajan A, Kim S, Gurdziel K, Mao G, Nangia-Makker P. et al. RAD6B Loss Disrupts Expression of Melanoma Phenotype in Part by Inhibiting WNT/β-Catenin Signaling. Am J Pathol. 2021;191:368-84
178. Xu J, Lv G, Xu B, Jiang B. Overexpression of UBE2M through Wnt/β-Catenin signaling is associated with poor prognosis and chemotherapy resistance in colorectal cancer. Transl Cancer Res. 2020;9:5614-25
179. Hu W, Li M, Chen Y, Gu X. UBE2S promotes the progression and Olaparib resistance of ovarian cancer through Wnt/β-catenin signaling pathway. J Ovarian Res. 2021;14:121
180. Chen J, Chen ZJ. Regulation of NF-κB by ubiquitination. Curr Opin Immunol. 2013;25:4-12
181. Deng L, Meng T, Chen L, Wei W, Wang P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct Target Ther. 2020;5:11
182. Zhang Y, Li Y, Yang X, Wang J, Wang R, Qian X. et al. Uev1A-Ubc13 catalyzes K63-linked ubiquitination of RHBDF2 to promote TACE maturation. Cell Signal. 2018;42:155-64
183. Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749-59
184. Xie H, He Y, Wu Y, Lu Q. Silencing of UBE2D1 inhibited cell migration in gastric cancer, decreasing ubiquitination of SMAD4. Infect Agent Cancer. 2021;16:63
185. Chen H, Hu Q, Wen T, Luo L, Liu L, Wang L. et al. Arteannuin B, a sesquiterpene lactone from Artemisia annua, attenuates inflammatory response by inhibiting the ubiquitin-conjugating enzyme UBE2D3-mediated NF-κB activation. Phytomedicine. 2024;124:155263
186. Gutschner T, Diederichs S. The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol. 2012;9:703-19
187. Razgonova MP, Zakharenko AM, Golokhvast KS, Thanasoula M, Sarandi E, Nikolouzakis K. et al. Telomerase and telomeres in aging theory and chronographic aging theory (Review). Mol Med Rep. 2020;22:1679-94
188. Robinson NJ, Schiemann WP. Telomerase in Cancer: Function, Regulation, and Clinical Translation. Cancers (Basel). 2022 14
189. Zalzman M, Meltzer WA, Portney BA, Brown RA, Gupta A. The Role of Ubiquitination and SUMOylation in Telomere Biology. Curr Issues Mol Biol. 2020;35:85-98
190. Sun ZW, Allis CD. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature. 2002;418:104-8
191. Cerone MA, Londono-Vallejo JA, Bacchetti S. Telomere maintenance by telomerase and by recombination can coexist in human cells. Hum Mol Genet. 2001;10:1945-52
192. Goubran HA, Kotb RR, Stakiw J, Emara ME, Burnouf T. Regulation of tumor growth and metastasis: the role of tumor microenvironment. Cancer Growth Metastasis. 2014;7:9-18
193. Shackelford RE, Kaufmann WK, Paules RS. Cell cycle control, checkpoint mechanisms, and genotoxic stress. Environ Health Perspect. 1999;107(Suppl 1):5-24
194. Chen L, Liu S, Tao Y. Regulating tumor suppressor genes: post-translational modifications. Signal Transduct Target Ther. 2020;5:90
195. Jesenberger V, Jentsch S. Deadly encounter: ubiquitin meets apoptosis. Nat Rev Mol Cell Biol. 2002;3:112-21
196. Lee SY, Ju MK, Jeon HM, Jeong EK, Lee YJ, Kim CH. et al. Regulation of Tumor Progression by Programmed Necrosis. Oxid Med Cell Longev. 2018;2018:3537471
197. Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta. 2016;1863:2977-92
198. Bergsagel PL, Chesi M. V. Molecular classification and risk stratification of myeloma. Hematol Oncol. 2013;31(Suppl 1):38-41
199. Xu Y, Zhang Z, Li J, Tong J, Cao B, Taylor P. et al. The ubiquitin-conjugating enzyme UBE2O modulates c-Maf stability and induces myeloma cell apoptosis. J Hematol Oncol. 2017;10:132
200. Wang X, Yin L, Yang L, Zheng Y, Liu S, Yang J. et al. Silencing ubiquitin-conjugating enzyme 2C inhibits proliferation and epithelial-mesenchymal transition in pancreatic ductal adenocarcinoma. Febs j. 2019;286:4889-909
201. Roman-Trufero M, Dillon N. The UBE2D ubiquitin conjugating enzymes: Potential regulatory hubs in development, disease and evolution. Front Cell Dev Biol. 2022;10:1058751
202. Zhang P, Guo H, Zhao F, Jia K, Yang F, Liu X. UBE2J1 knockdown promotes cell apoptosis in endometrial cancer via regulating PI3K/AKT and MDM2/p53 signaling. Open Med (Wars). 2023;18:20220567
203. Yang T, Pan Q, Yue R, Liu G, Zhou Y. Daphnetin alleviates silica-induced pulmonary inflammation and fibrosis by regulating the PI3K/AKT1 signaling pathway in mice. Int Immunopharmacol. 2024;133:112004
204. Zhou W, Xu J, Li H, Xu M, Chen ZJ, Wei W. et al. Neddylation E2 UBE2F Promotes the Survival of Lung Cancer Cells by Activating CRL5 to Degrade NOXA via the K11 Linkage. Clin Cancer Res. 2017;23:1104-16
205. Yan X, Chen D, Wang Y, Guo Y, Tong C, Wei J. et al. Identification of NOXA as a pivotal regulator of resistance to CAR T-cell therapy in B-cell malignancies. Signal Transduct Target Ther. 2022;7:98
206. Ashry R, Mustafa AM, Hausmann K, Linnebacher M, Strand S, Sippl W. et al. NOXA Accentuates Apoptosis Induction by a Novel Histone Deacetylase Inhibitor. Cancers (Basel). 2023 15
207. Mortezaee K. Immune escape: A critical hallmark in solid tumors. Life Sci. 2020;258:118110
208. Huang L, Liu H, Zhang K, Meng Q, Hu L, Zhang Y. et al. Ubiquitin-Conjugating Enzyme 2S Enhances Viral Replication by Inhibiting Type I IFN Production through Recruiting USP15 to Deubiquitinate TBK1. Cell Rep. 2020;32:108044
209. Peng Q, Chen Y, Zhao J, Zhu J, Zhuo H, Zheng Y. et al. RSK1 reprograms the ubiquitin pathway to promote immune suppression. Cell Rep. 2025;44:116323
210. Yue H, Wang J, Hou S, Zhang M. As a potential predictor of pan-cancer, UBE2S is related to tumor-associated macrophage infiltration. Future Oncol. 2023;19:1973-90
211. Wang B, Hu L, Zhang Z, Yin Y, Zheng L, Wu H. et al. UBE2S-mediated deubiquitination of GLUT1 via USP10 regulates glucose metabolic reprogramming and immune microenvironment to promote fibrosis in endometriosis. J Transl Med. 2025;23:1383
212. Rajabi M, Mousa SA. The Role of Angiogenesis in Cancer Treatment. Biomedicines. 2017 5
213. Basic VT, Jacobsen A, Sirsjö A, Abdel-Halim SM. TNF stimulation induces VHL overexpression and impairs angiogenic potential in skeletal muscle myocytes. Int J Mol Med. 2014;34:228-36
214. Shen R, Wu T, Huang P, Shao Q, Chen M. The clinicopathological significance of ubiquitin-conjugating enzyme E2C, leucine-rich repeated-containing G protein-coupled receptor, WW domain-containing oxidoreductase, and vasculogenic mimicry in invasive breast carcinoma. Medicine (Baltimore). 2019;98:e15232
215. Miskinyte S, Butler MG, Hervé D, Sarret C, Nicolino M, Petralia JD. et al. Loss of BRCC3 deubiquitinating enzyme leads to abnormal angiogenesis and is associated with syndromic moyamoya. Am J Hum Genet. 2011;88:718-28
216. Habu T, Harada KH. UBC13 is an RNF213-associated E2 ubiquitin-conjugating enzyme, and Lysine 63-linked ubiquitination by the RNF213-UBC13 axis is responsible for angiogenic activity. FASEB Bioadv. 2021;3:243-58
217. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020 368
218. Hosios AM, Manning BD. Cancer Signaling Drives Cancer Metabolism: AKT and the Warburg Effect. Cancer Res. 2021;81:4896-8
219. Wittekind C, Neid M. Cancer invasion and metastasis. Oncology. 2005;69(Suppl 1):14-6
220. Xu L, Dai B, Zhang L, Chen W, Rong S, Chen J. et al. UBE2C mediates follicular thyroid carcinoma invasion and metastasis via K29-Specific vimentin ubiquitination. Cancer Lett. 2025;617:217624
221. Chen M, Liu LX. MiR-525-5p Repressed Metastasis and Anoikis Resistance in Cervical Cancer via Blocking UBE2C/ZEB1/2 Signal Axis. Dig Dis Sci. 2020;65:2442-51
222. Qi S, Guan X, Zhang J, Yu D, Yu X, Li Q. et al. Targeting E2 ubiquitin-conjugating enzyme UbcH5c by small molecule inhibitor suppresses pancreatic cancer growth and metastasis. Mol Cancer. 2022;21:70
223. Hu W, Xiao L, Cao C, Hua S, Wu D. UBE2T promotes nasopharyngeal carcinoma cell proliferation, invasion, and metastasis by activating the AKT/GSK3β/β-catenin pathway. Oncotarget. 2016;7:15161-72
224. Akkız H, Şimşek H, Balcı D, Ülger Y, Onan E, Akçaer N. et al. Inflammation and cancer: molecular mechanisms and clinical consequences. Front Oncol. 2025;15:1564572
225. Gu Q, Zou J, Zhou Y, Deng Q. Mechanism of inflammasomes in cancer and targeted therapies. Front Oncol. 2023;13:1133013
226. Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat Immunol. 2011;12:715-23
227. Walsh MC, Lee J, Choi Y. Tumor necrosis factor receptor- associated factor 6 (TRAF6) regulation of development, function, and homeostasis of the immune system. Immunol Rev. 2015;266:72-92
228. Deng L, Wang C, Spencer E, Yang L, Braun A, You J. et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351-61
229. Zhang H, Hu H, Greeley N, Jin J, Matthews AJ, Ohashi E. et al. STAT3 restrains RANK- and TLR4-mediated signalling by suppressing expression of the E2 ubiquitin-conjugating enzyme Ubc13. Nat Commun. 2014;5:5798
230. Roth TL, Marson A. Genetic Disease and Therapy. Annu Rev Pathol. 2021;16:145-66
231. Qi Z, Zhang Y, Qi S, Ling L, Gui L, Yan L. et al. Salidroside Inhibits HMGB1 Acetylation and Release through Upregulation of SirT1 during Inflammation. Oxid Med Cell Longev. 2017;2017:9821543
232. Qi X, Wang H, Xia L, Lin R, Li T, Guan C. et al. miR-30b-5p releases HMGB1 via UBE2D2/KAT2B/HMGB1 pathway to promote pro-inflammatory polarization and recruitment of macrophages. Atherosclerosis. 2021;324:38-45
233. Pikor L, Thu K, Vucic E, Lam W. The detection and implication of genome instability in cancer. Cancer Metastasis Rev. 2013;32:341-52
234. Torgovnick A, Schumacher B. DNA repair mechanisms in cancer development and therapy. Front Genet. 2015;6:157
235. Salmaninejad A, Ilkhani K, Marzban H, Navashenaq JG, Rahimirad S, Radnia F. et al. Genomic Instability in Cancer: Molecular Mechanisms and Therapeutic Potentials. Curr Pharm Des. 2021;27:3161-9
236. Bergink S, Jentsch S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature. 2009;458:461-7
237. Chen S, Wang C, Sun L, Wang DL, Chen L, Huang Z. et al. RAD6 promotes homologous recombination repair by activating the autophagy-mediated degradation of heterochromatin protein HP1. Mol Cell Biol. 2015;35:406-16
238. Haddad DM, Vilain S, Vos M, Esposito G, Matta S, Kalscheuer VM. et al. Mutations in the intellectual disability gene Ube2a cause neuronal dysfunction and impair parkin-dependent mitophagy. Mol Cell. 2013;50:831-43
239. Han X, Zhang L, Chung J, Mayca Pozo F, Tran A, Seachrist DD. et al. UbcH7 regulates 53BP1 stability and DSB repair. Proc Natl Acad Sci U S A. 2014;111:17456-61
240. Gemoll T, Miroll E, Klein O, Lischka A, Eravci M, Thorns C. et al. Spatial UBE2N protein expression indicates genomic instability in colorectal cancers. BMC Cancer. 2019;19:710
241. Hu L, Li X, Liu Q, Xu J, Ge H, Wang Z. et al. UBE2S, a novel substrate of Akt1, associates with Ku70 and regulates DNA repair and glioblastoma multiforme resistance to chemotherapy. Oncogene. 2017;36:1145-56
242. He Z, Wilson A, Rich F, Kenwright D, Stevens A, Low YS. et al. Chromosomal instability and its effect on cell lines. Cancer Rep (Hoboken). 2023;6:e1822
243. Shoshani O, Bakker B, de Haan L, Tijhuis AE, Wang Y, Kim DH. et al. Transient genomic instability drives tumorigenesis through accelerated clonal evolution. Genes Dev. 2021;35:1093-108
244. Fraile JM, Quesada V, Rodríguez D, Freije JM, López-Otín C. Deubiquitinases in cancer: new functions and therapeutic options. Oncogene. 2012;31:2373-88
245. Ikeda F. Ubiquitin conjugating enzymes in the regulation of the autophagy-dependent degradation pathway. Matrix Biol. 2021;100-101:23-9
246. Wiener R, DiBello AT, Lombardi PM, Guzzo CM, Zhang X, Matunis MJ. et al. E2 ubiquitin-conjugating enzymes regulate the deubiquitinating activity of OTUB1. Nat Struct Mol Biol. 2013;20:1033-9
247. Burroughs AM, Jaffee M, Iyer LM, Aravind L. Anatomy of the E2 ligase fold: implications for enzymology and evolution of ubiquitin/Ub-like protein conjugation. J Struct Biol. 2008;162:205-18
248. Kar G, Keskin O, Nussinov R, Gursoy A. Human proteome-scale structural modeling of E2-E3 interactions exploiting interface motifs. J Proteome Res. 2012;11:1196-207
249. Streich FC Jr, Lima CD. Structural and functional insights to ubiquitin-like protein conjugation. Annu Rev Biophys. 2014;43:357-79
250. Cook BW, Shaw GS. Architecture of the catalytic HPN motif is conserved in all E2 conjugating enzymes. Biochem J. 2012;445:167-74
251. Gao C, Liu YJ, Yu J, Wang R, Shi JJ, Chen RY. et al. Unraveling the Role of Ubiquitin-Conjugating Enzyme UBE2T in Tumorigenesis: A Comprehensive Review. Cells. 2024 14
252. Zhu Z, Cao C, Zhang D, Zhang Z, Liu L, Wu D. et al. UBE2T-mediated Akt ubiquitination and Akt/β-catenin activation promotes hepatocellular carcinoma development by increasing pyrimidine metabolism. Cell Death Dis. 2022;13:154
253. Alpi AF, Chaugule V, Walden H. Mechanism and disease association of E2-conjugating enzymes: lessons from UBE2T and UBE2L3. Biochem J. 2016;473:3401-19
254. Ho NPY, Leung CON, Wong TL, Lau EYT, Lei MML, Mok EHK. et al. The interplay of UBE2T and Mule in regulating Wnt/β-catenin activation to promote hepatocellular carcinoma progression. Cell Death Dis. 2021;12:148
255. Alpi AF, Pace PE, Babu MM, Patel KJ. Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol Cell. 2008;32:767-77
256. Miles JA, Frost MG, Carroll E, Rowe ML, Howard MJ, Sidhu A. et al. The Fanconi Anemia DNA Repair Pathway Is Regulated by an Interaction between Ubiquitin and the E2-like Fold Domain of FANCL. J Biol Chem. 2015;290:20995-1006
257. Lin Y, Hwang WC, Basavappa R. Structural and functional analysis of the human mitotic-specific ubiquitin-conjugating enzyme, UbcH10. J Biol Chem. 2002;277:21913-21
258. Olsen SK, Lima CD. Structure of a ubiquitin E1-E2 complex: insights to E1-E2 thioester transfer. Mol Cell. 2013;49:884-96
259. Nameki N, Yoneyama M, Koshiba S, Tochio N, Inoue M, Seki E. et al. Solution structure of the RWD domain of the mouse GCN2 protein. Protein Sci. 2004;13:2089-100
260. Xie C, Powell C, Yao M, Wu J, Dong Q. Ubiquitin-conjugating enzyme E2C: a potential cancer biomarker. Int J Biochem Cell Biol. 2014;47:113-7
261. Rape M, Kirschner MW. Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature. 2004;432:588-95
262. Eldridge AG, O'Brien T. Therapeutic strategies within the ubiquitin proteasome system. Cell Death Differ. 2010;17:4-13
263. Barreyro L, Sampson AM, Ishikawa C, Hueneman KM, Choi K, Pujato MA. et al. Blocking UBE2N abrogates oncogenic immune signaling in acute myeloid leukemia. Sci Transl Med. 2022;14:eabb7695
264. Schwalen F, Ghadi C, Ibazizene L, Khan SU, Sopkova-de Oliveira Santos J, Weiswald LB. et al. UBE2N: Hope on the Cancer Front, How to Inhibit This Promising Target Prospect? J Med Chem. 2025;68:915-28
265. Hussein HA, Borrel A, Geneix C, Petitjean M, Regad L, Camproux AC. PockDrug-Server: a new web server for predicting pocket druggability on holo and apo proteins. Nucleic Acids Res. 2015;43:W436-42
266. Hodge CD, Edwards RA, Markin CJ, McDonald D, Pulvino M, Huen MS. et al. Covalent Inhibition of Ubc13 Affects Ubiquitin Signaling and Reveals Active Site Elements Important for Targeting. ACS Chem Biol. 2015;10:1718-28
267. Rout MK, Hodge CD, Markin CJ, Xu X, Glover JN, Xiao W. et al. Stochastic gate dynamics regulate the catalytic activity of ubiquitination enzymes. J Am Chem Soc. 2014;136:17446-58
268. Chua MD, Mineva GM, Guttman JA. Ube2N is present and functions within listeria Actin-rich structures and lamellipodia: A localization and pharmacological inhibition study. Anat Rec (Hoboken). 2023;306:1140-8
269. Dikshit A, Jin YJ, Degan S, Hwang J, Foster MW, Li CY. et al. UBE2N Promotes Melanoma Growth via MEK/FRA1/SOX10 Signaling. Cancer Res. 2018;78:6462-72
270. Du X, Song H, Shen N, Hua R, Yang G. The Molecular Basis of Ubiquitin-Conjugating Enzymes (E2s) as a Potential Target for Cancer Therapy. Int J Mol Sci. 2021 22
271. Cheng SS, Yang GJ, Wang W, Leung CH, Ma DL. The design and development of covalent protein-protein interaction inhibitors for cancer treatment. J Hematol Oncol. 2020;13:26
272. Engel J, Richters A, Getlik M, Tomassi S, Keul M, Termathe M. et al. Targeting Drug Resistance in EGFR with Covalent Inhibitors: A Structure-Based Design Approach. J Med Chem. 2015;58:6844-63
273. Sutanto F, Konstantinidou M, Dömling A. Covalent inhibitors: a rational approach to drug discovery. RSC Med Chem. 2020;11:876-84
274. Rauert-Wunderlich H, Siegmund D, Maier E, Giner T, Bargou RC, Wajant H. et al. The IKK inhibitor Bay 11-7082 induces cell death independent from inhibition of activation of NFκB transcription factors. PLoS One. 2013;8:e59292
275. Scheper J, Guerra-Rebollo M, Sanclimens G, Moure A, Masip I, González-Ruiz D. et al. Protein-protein interaction antagonists as novel inhibitors of non-canonical polyubiquitylation. PLoS One. 2010;5:e11403
276. Ardecky R, Madiraju C, Matsuzawa SI, Zou J, Ganji S, Pass I. et al. Selective UBC 13 Inhibitors. Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD): National Center for Biotechnology Information (US). 2010
277. Ceccarelli DF, Tang X, Pelletier B, Orlicky S, Xie W, Plantevin V. et al. An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell. 2011;145:1075-87
278. Lu H, Zhou Q, He J, Jiang Z, Peng C, Tong R. et al. Recent advances in the development of protein-protein interactions modulators: mechanisms and clinical trials. Signal Transduct Target Ther. 2020;5:213
279. Brenke JK, Popowicz GM, Schorpp K, Rothenaigner I, Roesner M, Meininger I. et al. Targeting TRAF6 E3 ligase activity with a small-molecule inhibitor combats autoimmunity. J Biol Chem. 2018;293:13191-203
280. Lu J, Gu X, Liu F, Rui Z, Liu M, Zhao L. Antitumor effects of hsa-miR661-3p on non-small cell lung cancer in vivo and in vitro. Oncol Rep. 2019;41:2987-96
281. Wei X, You X, Zhang J, Zhou C. MicroRNA-1305 Inhibits the Stemness of LCSCs and Tumorigenesis by Repressing the UBE2T-Dependent Akt-Signaling Pathway. Mol Ther Nucleic Acids. 2019;16:721-32
282. Pal A, Young MA, Donato NJ. Emerging potential of therapeutic targeting of ubiquitin-specific proteases in the treatment of cancer. Cancer Res. 2014;74:4955-66
283. Sanders MA, Brahemi G, Nangia-Makker P, Balan V, Morelli M, Kothayer H. et al. Novel inhibitors of Rad6 ubiquitin conjugating enzyme: design, synthesis, identification, and functional characterization. Mol Cancer Ther. 2013;12:373-83
284. Kothayer H, Spencer SM, Tripathi K, Westwell AD, Palle K. Synthesis and in vitro anticancer evaluation of some 4,6-diamino-1,3,5-triazine-2-carbohydrazides as Rad6 ubiquitin conjugating enzyme inhibitors. Bioorg Med Chem Lett. 2016;26:2030-4
285. King EA, Cho Y, Hsu NS, Dovala D, McKenna JM, Tallarico JA. et al. Chemoproteomics-enabled discovery of a covalent molecular glue degrader targeting NF-κB. Cell Chem Biol. 2023;30:394-402.e9
286. Huang M, Zhou Y, Duan D, Yang C, Zhou Z, Li F. et al. Targeting ubiquitin conjugating enzyme UbcH5b by a triterpenoid PC3-15 from Schisandra plants sensitizes triple-negative breast cancer cells to lapatinib. Cancer Lett. 2021;504:125-36
287. Xu T, Ma Q, Li Y, Yu Q, Pan P, Zheng Y. et al. A small molecule inhibitor of the UBE2F-CRL5 axis induces apoptosis and radiosensitization in lung cancer. Signal Transduct Target Ther. 2022;7:354
288. Wang C, Shi G, Ji X. Design, synthesis, and anticancer activity evaluation of irreversible allosteric inhibitors of the ubiquitin-conjugating enzyme Ube2g2. Medchemcomm. 2018;9:1818-25
289. Zhou P, Chen X, Li M, Tan J, Zhang Y, Yuan W. et al. 2-D08 as a SUMOylation inhibitor induced ROS accumulation mediates apoptosis of acute myeloid leukemia cells possibly through the deSUMOylation of NOX2. Biochem Biophys Res Commun. 2019;513:1063-9
290. Cheng J, Fan YH, Xu X, Zhang H, Dou J, Tang Y. et al. A small-molecule inhibitor of UBE2N induces neuroblastoma cell death via activation of p53 and JNK pathways. Cell Death Dis. 2014;5:e1079
291. Cornwell MJ, Thomson GJ, Coates J, Belotserkovskaya R, Waddell ID, Jackson SP. et al. Small-Molecule Inhibition of UBE2T/FANCL-Mediated Ubiquitylation in the Fanconi Anemia Pathway. ACS Chem Biol. 2019;14:2148-54
292. Yu Z, Jiang X, Qin L, Deng H, Wang J, Ren W. et al. A novel UBE2T inhibitor suppresses Wnt/β-catenin signaling hyperactivation and gastric cancer progression by blocking RACK1 ubiquitination. Oncogene. 2021;40:1027-42
293. Yan Y, Nie Y, Peng C, Xing F, Ji S, Liu H. et al. The circular RNA hsa_circ_0001394 promotes hepatocellular carcinoma progression by targeting the miR-527/UBE2A axis. Cell Death Discov. 2022;8:81
294. Cheng CM, Shiah SG, Huang CC, Hsiao JR, Chang JY. Up-regulation of miR-455-5p by the TGF-β-SMAD signalling axis promotes the proliferation of oral squamous cancer cells by targeting UBE2B. J Pathol. 2016;240:38-49
295. Zhang Y, Han T, Wei G, Wang Y. Inhibition of microRNA-17/20a suppresses cell proliferation in gastric cancer by modulating UBE2C expression. Oncol Rep. 2015;33:2529-36
296. Hu J, Wu X, Yang C, Rashid K, Ma C, Hu M. et al. Anticancer effect of icaritin on prostate cancer via regulating miR-381-3p and its target gene UBE2C. Cancer Med. 2019;8:7833-45
297. Huang W, Zhou Y, Ren J, Chen C. UBE2C, targeted by miR-140-3p, promotes the progression of osteosarcoma via PI3K/AKT signaling pathway. J Recept Signal Transduct Res. 2024;44:107-14
298. Li W, Wang F, Wang X, Xu W, Liu F, Hu R. et al. Curcumin inhibits prostate cancer by upregulating miR-483-3p and inhibiting UBE2C. J Biochem Mol Toxicol. 2024;38:e23645
299. Yen MC, Wu KL, Liu YW, Chang YY, Chang CY, Hung JY. et al. Ubiquitin Conjugating Enzyme E2 H (UBE2H) Is Linked to Poor Outcomes and Metastasis in Lung Adenocarcinoma. Biology (Basel). 2021 10
300. Manvati S, Mangalhara KC, Kalaiarasan P, Srivastava N, Kumar B, Bamezai RN. MiR-101 induces senescence and prevents apoptosis in the background of DNA damage in MCF7 cells. PLoS One. 2014;9:e111177
301. Zhao Z, Tan X, Zhao A, Zhu L, Yin B, Yuan J. et al. microRNA-214-mediated UBC9 expression in glioma. BMB Rep. 2012;45:641-6
302. Gu Y, Feng X, Jin Y, Liu Y, Zeng L, Zhou D. et al. Upregulation of miRNA-10a-5p promotes tumor progression in cervical cancer by suppressing UBE2I signaling. J Obstet Gynaecol. 2023;43:2171283
303. Guo Z, Wang Y, Qin W, Heng Y, Chen X, Liu N. et al. miR-122-3p targets UBE2I to regulate the immunosuppression of liver cancer and the intervention of Liujunzi formula. J Ethnopharmacol. 2024;329:118081
304. Zhang E, Liu Q, Wang Y, Wang H, He L, Jin X. et al. MicroRNA miR-147b promotes tumor growth via targeting UBE2N in hepatocellular carcinoma. Oncotarget. 2017;8:114072-80
305. Song TT, Xu F, Wang W. Inhibiting ubiquitin conjugating enzyme E2 N by microRNA-590-3p reduced cell growth of cervical carcinoma. Kaohsiung J Med Sci. 2020;36:501-7
306. Vallabhaneni KC, Penfornis P, Xing F, Hassler Y, Adams KV, Mo YY. et al. Stromal cell extracellular vesicular cargo mediated regulation of breast cancer cell metastasis via ubiquitin conjugating enzyme E2 N pathway. Oncotarget. 2017;8:109861-76
307. Li L, Li Q. miR-543 impairs breast cancer cell phenotypes by targeting and suppressing ubiquitin-conjugating enzyme E2T (UBE2T). Bioengineered. 2021;12:12394-406
308. Ren X, Li A, Ying E, Fang J, Li M, Yu J. Upregulation of ubiquitin-conjugating enzyme E2T (UBE2T) predicts poor prognosis and promotes hepatocellular carcinoma progression. Bioengineered. 2021;12:1530-42
309. Cao W, Ni L, Li P, Wang QX, Li MM, Huang SH. et al. miR-498 Targets UBE2T to Inhibit the Proliferation of Malignant Melanoma Cells. Technol Cancer Res Treat. 2022;21:15330338221082431
310. Wu Y, Zhang C, Peng D, He S, Huang C, Qian J. et al. MiR-182-5p inhibits the tumorigenesis of clear cell renal cell carcinoma by repressing UBE2T. Hum Cell. 2022;35:542-56
311. Wang L, Zhang Z, Tian H. Hsa_circ_0092887 targeting miR-490-5p/UBE2T promotes paclitaxel resistance in non-small cell lung cancer. J Clin Lab Anal. 2023;37:e24781
312. Chen Z, Jin P, Chen Z, Ye F, Ren Z, Ji T. et al. The expression of circ_0090049 in hepatocellular carcinoma and the molecular regulation mechanism of other biological functions. Anticancer Drugs. 2022;33:48-60
313. Koo N, Sharma AK, Narayan S. Therapeutics Targeting p53-MDM2 Interaction to Induce Cancer Cell Death. Int J Mol Sci. 2022 23
314. Mayca Pozo F, Tang J, Bonk KW, Keri RA, Yao X, Zhang Y. Regulatory cross-talk determines the cellular levels of 53BP1 protein, a critical factor in DNA repair. J Biol Chem. 2017;292:5992-6003
315. Ma X, Qi W, Yang F, Pan H. UBE2L3 promotes lung adenocarcinoma invasion and metastasis through the GSK-3β/Snail signaling pathway. Am J Transl Res. 2022;14:4549-61
316. Gurnari C, Voso MT, Girardi K, Mastronuzzi A, Strocchio L. Acute Promyelocytic Leukemia in Children: A Model of Precision Medicine and Chemotherapy-Free Therapy. Int J Mol Sci. 2021 22
317. Wang C, Zhang Y, Chen W, Wu Y, Xing D. New-generation advanced PROTACs as potential therapeutic agents in cancer therapy. Mol Cancer. 2024;23:110
318. Li X, Song Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J Hematol Oncol. 2020;13:50
319. Sasso JM, Tenchov R, Wang D, Johnson LS, Wang X, Zhou QA. Molecular Glues: The Adhesive Connecting Targeted Protein Degradation to the Clinic. Biochemistry. 2023;62:601-23
Author contact
![]() Corresponding authors: guanxqorg.cn (Xiaoqing Guan); jqinac.cn (Jiang-jiang Qin) and kaimiaoedu.mo (Kai Miao).
Corresponding authors: guanxqorg.cn (Xiaoqing Guan); jqinac.cn (Jiang-jiang Qin) and kaimiaoedu.mo (Kai Miao).