Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(6):804-814. doi:10.7150/ijbs.19398 This issue Cite
Research Paper
Stroma-derived Fibrinogen-like Protein 2 Activates Cancer-associated Fibroblasts to Promote Tumor Growth in Lung Cancer
Ying Zhu1, 2*, Longhui Zhang1, 2*, Haoran Zha1, 2, Fei Yang3, Chunyan Hu1, 2, Lin Chen4, Bo Guo3 ![]() , Bo Zhu1, 2
, Bo Zhu1, 2 ![]()
1. Institute of Cancer, Xinqiao Hospital, Third Military Medical University, Chongqing 400037, P. R. China;
2. Chongqing Key Laboratory of Immunotherapy, Chongqing 400037, P. R. China;
3. Department of Pathogenic Biology, Third Military Medical University, Chongqing, 400038, P. R. China;
4. Department of Rehabilitation Medicine, Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing, 400042, P. R. China
* These authors contributed equally to this work.
Received 2017-1-30; Accepted 2017-3-10; Published 2017-6-1
Abstract

Fibrinogen-like protein 2 (Fgl2), a member of the fibrinogen super family, is a pleiotropic cytokine that impacts diverse cellular functions. Previous studies have shown that tumor cell-derived Fgl2 promotes tumorigenesis and metastasis in immune-deficient mice, and it also functions as an immune-suppressive modulator in glioblastoma multiform (GMB). This study aimed to evaluate whether and how tumor stroma-derived Fgl2 affects tumorigenesis and tumor progression. We established the syngeneic transplantable Lewis lung carcinoma (LLC) model in Fgl2-knock-out (Fgl2-KO) mice and we found that deficiency of host Fgl2 is associated with reduced growth of syngeneic LLC tumors. Furthermore, we confirmed that host Fgl2 deficiency significantly decreased the accumulation of myeloid-derived suppressor cells (MDSCs) through down-regulation of chemokine (C-X-C motif) ligand 12 (CXCL12) expression. More importantly, we demonstrated that Fgl2 induced an activated and pro-tumorigenic phenotype of cancer-associated fibroblasts (CAFs) which are the principal source of CXCL12 in the tumor microenvironment (TME). Our results present a novel role of stroma-derived Fgl2 in CAF activation and function, suggesting that Fgl2 is an effective therapeutic target for treating lung cancer.
Keywords: Fgl2, cancer-associated fibroblast, myeloid-derived suppressor cell, lung cancer.
Introduction
Tumor cells have a close interaction with their surrounding cells, including endothelial, fibroblastic and immune cells, to form the TME. The TME facilitates tumor cell growth and escape from host immune surveillance by providing a chronic inflammatory, immunosuppressive, and pro-angiogenic environment. As it is widely recognized that the TME plays a critical role in resistance to various therapies in the clinic, a better understanding of the TME would benefit clinical interventions against cancer [1, 2].
Fgl2, a member of the fibrinogen super family, possesses prothrombinase activity and immune regulatory functions in a variety of conditions including viral-induced inflammation, xenograft rejection, chronic obstructive pulmonary disease and abortion [3-6]. In the previous studies, Fgl2 has been showed to play a multimodal role in the innate and acquired immune system, acting as an effect molecular of Treg cells, suppressing T cell proliferation, inducing apoptosis in B cells, promoting macrophage activation and inhibiting maturation of dendritic cells (DCs) [4, 7-9]. Previous studies have shown that Fgl2 is markedly up-regulated in tumors and promotes tumorigenesis and metastasis in immune-deficient mice [10-12]. In addition, a recent study in GBM showed that glioma cell-derived Fgl2 functions as an immune-suppressive modulator by upregulating the expression levels of CD39 and PD-1, as well as increasing the frequency of immunosuppressive cells [13]. However, these studies focused on tumor cell-derived Fgl2; the functions of tumor stroma-derived Fgl2 in the antitumor immunity process are still unknown.
Moreover, Fgl2 has shown a trend toward co-occurrence with Foxp3 in human malignancies, indicating that tumor stroma-derived Fgl2 may play an essential role in tumor progression [14]. Therefore, we established a syngeneic transplantable lung cancer model in Fgl2-KO mice to clarify the function of Fgl2 derived from tumor stroma in tumor progression, and we found that stroma-derived Fgl2 enhanced the CAF phenotype and function to promote tumorigenesis and tumor progression in lung cancer.
Materials and Methods
Mice
Fgl2-KO mice were kindly provided by Dr. Steve Smiley (The Trudeau Institute, NY, USA). The mice were kept under specific pathogen-free conditions at the Animal Center of the Third Military Medical University, and their homozygous wild-type (WT) littermates were used as controls. C57BL/6 mice were obtained from the Animal Institute of the Academy of Medical Science (Beijing, China). All animal experiments were approved by the ethics committee of the Third Military Medical University.
Cell lines and cell culture
LLC cells (Lewis lung carcinoma cells) were obtained from the American Type Culture Collection (ATCC). The cells were cultured in DMEM medium (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco), 2 mM fresh L-glutamine and 100 units/ml penicillin/streptomycin (HyClone) and were incubated in a 37°C incubator under 5% CO2.
Proliferation assay
Cell Counting Kit-8 (Beyotime Biotechnology) was used following the manufacturer's instructions. Briefly, 5×103 LLC cells were cultured in 100 µl of DMEM in 96-well culture plates. In addition, LLC cells were treated with or without mouse recombinant Fgl2 (1 µg/ml or 5 µg/ml, R&D Systems) for 24, 48 or 72 hours. Then, 100 µl of CCK-8 reagent was added to the cells and cultured for 1 hour in a 37°C incubator under 5% CO2. Finally, luminescence was measured using a Varioskan Flash microplate reader.
Apoptosis assay
An Annexin V Apoptosis Detection Kit with 7-AAD (Biolegend) was used following the manufacturer's instructions. In brief, 2×105 LLC cells were cultured in 250 µl DMEM in 24-well culture plates. In addition, LLC cells were treated with or without recombinant Fgl2 (1 µg/ml or 5 µg/ml, R&D Systems) for 24 or 48 hours. Cells were harvested at the indicated time point and stained with FITC-Annexin V and 7AAD at a dilution of 1:20. The cells were incubated for 15 min at room temperature in the dark. Samples were acquired on a Canto II flow cytometer (BD Biosciences) and the data were analyzed with FlowJo software.
Tumor challenge experiments
LLC cells were cultured until they reached 70% confluence. Then, Fgl2-KO and WT mice (male or female, 6-8 weeks old) were injected subcutaneously in the right lateral flank with 5×105 LLC cells in 100 µl PBS. Tumor sizes were measured using calipers every three days till sacrifice and tumor volume was calculated by the following formula: volume = 0.5 × (width) 2 × length.
Tumor digestion, antibody staining of cell suspensions and flow cytometry analysis
Single-cell suspensions of transplanted tumors were obtained by digestion with 1 mg/ml Collagenase I (Sigma) and 1 mg/ml Dispase II (Roche) in RPMI containing 5 % FBS for 45 min at 37°C under shaking condition. The cell suspension was passed through a 70 μm nylon mesh and centrifuged. After red blood cell lysis, Fc receptors were blocked with anti-mouse FcR antibody (clone 93, Biolegend) for 15 min at a 1:100 dilution on ice. Then, cells were stained with anti-CD45 (clone 30-F11, Biolegend), CD11b (clone M1/70, Biolegend), Gr-1 (clone RB6-8C5, Biolegend), F4/80 (clone BM8, Biolegend), PDGFRα (clone APA5; Biolegend), CD206 (clone C068C2, Biolegend), CD11c (clone N418, Biolegend) and MHC II (I-A/I-E, clone M5/114.15.2, Biolegend) antibodies for 30 min at a 1:100 dilution on ice. For analysis of Treg cells in the tumor, cells were stained with anti-CD4 (clone GK1.5, Biolegend, 1:100), Foxp3 (clone FJK-16s, eBioscience, 1:50) antibodies using a Foxp3 staining buffer kit following the instructions (eBioscience). Samples were acquired on a Canto II flow cytometer (BD Biosciences) and the data were analyzed with FlowJo software.
Isolation and stimulation of CAFs
Transplanted tumors were harvested at the indicated time points to prepare single-cell suspensions as previously described. After red blood cell lysis and blocking of Fc receptors, cells were stained with anti-PDGFRα and anti-F4/80 antibodies (Biolegend) at a 1:100 dilution for 30 min on ice. CAFs (PDGFRα+F4/80-) were sorted by Fluorescence Activated Cell Sorting (FACS) on an Aria II cell sorter (BD Biosciences) [15]. Sorted CAFs were suspended in RNA lysis buffer (Invitrogen) or seeded in 24-well plates in DMEM containing 10% FBS and 100 units/ml penicillin/streptomycin. Subsequently, non-adherent cells were removed by extensive washing with PBS and adherent cells were treated with recombinant mouse Fgl2 (1 µg/ml or 5 µg/ml, R&D Systems) for further analysis.
RNA isolation and qRT-PCR
Total RNA was extracted from tumors using TRIzol reagent (Takara). Furthermore, total RNA of CAFs from WT and Fgl2-KO mice was extracted using Micro Total RNA Isolation Kit (Invitrogen). RNA was reverse-transcribed with Reverse Transcription Reagent (Takara), following the manufacturer's instructions. The expression levels of genes of interest were measured using an ABI 7500 Real-time PCR machine and a SYBR Premix Ex TaqTM kit (Takara). The sequences of the primer pairs used are provided in the supplementary section
Western blot analysis
Proteins of CAFs were extracted by RIPA buffer (Beyotime Biotechnology) with 1% PMSF (Beyotime Biotechnology). Then, a BCA kit (Beyotime Biotechnology) was used to examine the protein concentrations. Equal protein amounts of samples were resolved on a 12% SDS-PAGE gel and transferred onto a polyvinylidene difluoride (PVDF) membrane. After blocking with 5% nonfat milk for 1 hour at 37°C, the membrane was applied with the primary antibodies overnight at 4°C: rabbit anti-αSMA antibody (Abcam, 1:1000) and mouse anti-α-tubulin antibody (Sigma, 1:1000). After being washed with 0.1% TBST, goat anti-rabbit secondary antibody (Beyotime Biotechnology, 1:5000) and goat anti-mouse secondary antibody (Beyotime Biotechnology, 1:5000) were applied, followed by detection of the chemiluminescence signal (Pierce).
Gene expression data from cBioportal for Cancer Genomics
Human non-small cell lung cancer (NSCLC) gene expression data were obtained from the Cancer Genome Atlas (TCGA) portal (http://www.cbioportal.org/Accessed December 3, 2016). Pearson's correlation coefficients were calculated to compare the Fgl2 gene and immune-related genes/cytokines expression using R statistical computing [16, 17].
Statistical analysis
Statistical analysis data are expressed as the mean ± SEM. Significance was assessed by two-tailed unpaired Student's t-test or other statistical method indicated in the test. Statistical significance is indicated as *p< 0.05, **p< 0.01 and ***p< 0.001. Statistical calculations were performed using GraphPad Prism 7.0 software. All of the experiments were independently repeated at least 3 times.
Results
Deficiency of host Fgl2 reduces growth of transplanted lung cancer
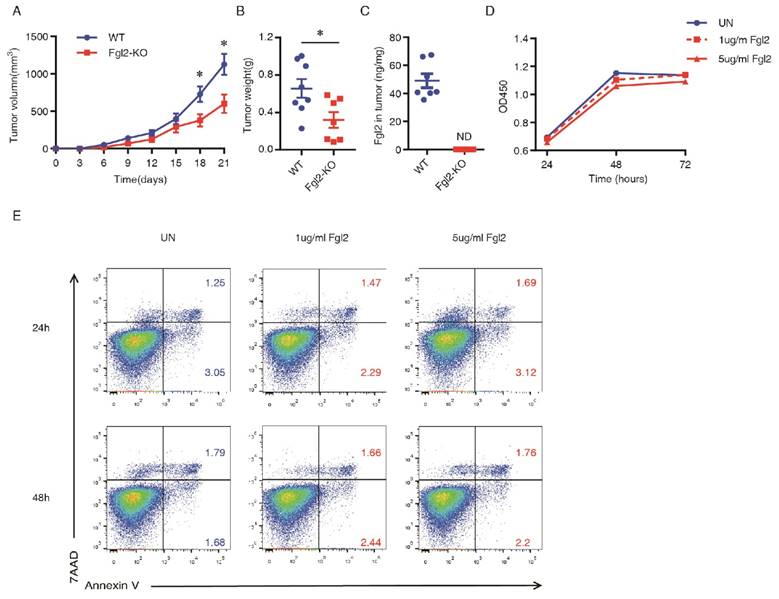
Previous studies have demonstrated that Fgl2 in tumor cells plays an important role in promoting tumor progression in hepatocellular carcinoma, colorectal cancer and gliomas [12, 13, 18]. To investigate whether Fgl2 was widely produced by various cancer cells in addition to glioma and hepatoma cells, we analyzed the mRNA levels of Fgl2 in different types of cancer cell lines using the Cancer Cell Line Encyclopedia (CCLE) database (http://www.broadinstitute.org/ccle/home). Unexpectedly, we noted that Fgl2 was barely expressed by epithelial cancer cells (Supplementary Figure 1). Therefore, we decided to explore the role of Fgl2 in cancer progression by using a genetic Fgl2 deficient mouse model, which might be more relevant to the actual state of human malignances. We injected LLC cells subcutaneously into Fgl2-KO and WT mice. As shown, the rate of tumor growth in the Fgl2-KO mice was slowed compared with WT mice (Figure 1A). Additionally, the weights of excised tumors at termination (day 21) were also markedly smaller in Fgl2-KO mice (Figure 1B). Moreover, we found that Fgl2 was not detected in the lysates of LLC implanted tumors from Fgl2-KO mice by ELISA (Figure 1C), indicating that LLC tumor cells do not secrete Fgl2 in the TME. To further confirm this finding, four populations of cells (tumor cells, CD45-PDGFRα-; cancer-associated fibroblasts (CAFs), CD45-PDGFRα+; myeloid cells, CD45+CD11b+; lymphocytes, CD45+CD11b-) were sorted from LLC implanted tumors in C57BL/6 mice by FACS (Supplementary Figure 2A). Fgl2 levels in these four populations of cells from TME and LLC cells cultured in vitro were detected by ELISA. As shown, myeloid cells and lymphocytes were the main sources of Fgl2 in the TME whereas tumor cells sorted from implanted tumors and LLC cells cultured in vitro were barely expressed Fgl2 (Supplementary Figure 2B). The above data strongly suggested that tumor stroma-derived Fgl2 promotes tumor growth in the LLC implanted tumor model. To determine whether Fgl2 influences tumor cells directly, we assessed the effect of Fgl2 on proliferation and apoptosis of LLC cells in vitro. After being treated with recombinant Fgl2 (1 µg/ml or 5 µg/ml), the proliferation and apoptosis rate of LLC cells was not affected (Figure 1D&E). These findings implied that Fgl2 expressed by tumor stroma promotes lung carcinoma growth by modeling the pro-tumorigenic microenvironment but does not regulate tumor cells directly.
Deficiency of Fgl2 expression in stromal cells reduces growth of a syngeneic murine transplantable lung carcinoma. Briefly, 5 x 105 LLC cells were subcutaneously injected in WT or Fgl2-KO mice (n = 7 or 8). Tumor volumes were measured and shown versus time (A). Tumor weight was measured when mice were sacrificed at day 21 (B). The level of Fgl2 in tumors of WT and Fgl2-KO mice was detected by ELISA (C). LLC cells were treated with Fgl2 (1 µg/ml or 5 µg/ml) for 24, 48 and 96 hours. The proliferation of LLC cells was measured by a CCK8 assay, and the absorbance at 450 nm was detected (D). The apoptosis of LLC cells was examined using Annexin V and 7AAD staining (E). The data represent means ± SEM. *p<0.05, **p<0.01, ***p<0.001 (A&E, ANOVA followed by Sitak's test for multiple comparisons; B&C, two-tailed unpaired Student's t-test) versus the indicated comparisons.
Fgl2 is required for the infiltration of MDSCs in syngeneic transplanted lung tumors
We have found that deficiency of stromal Fgl2 reduced tumor growth, so we determined to figure out the stromal alteration responsible for this effect. In a murine gliomas model, investigators have found that Fgl2 augments immunosuppression in the TME with increased immune-suppressive cells [13]. As the tumor-promoting effects of immune suppressive cells such as MDSCs, TAMs and Treg cells have been well investigated in lung carcinoma development, we suspected that Fgl2 may promote LLC tumor growth by recruitment of immune-suppressive cells [19-21]. The transplanted tumors from WT and Fgl2-KO mice were harvested and digested, further analyzed the cell suspension by flow cytometry (Figure 2A-E). In addition, we harvested the tumors 15 days after implanted, when the tumor volumes of the two genotypes of mice were nearly the same, demonstrating that the differences in the composition of tumors were not due to smaller tumor volume in Fgl2-KO mice. The results showed that the proportion of total leukocytes (CD45+) within tumor tissues was not significantly different in Fgl2-KO mice compared with WT mice (Figure 2A); whereas the percentage of MDSCs (CD11b+Gr-1+) was significantly reduced in Fgl2-KO mice compared with WT mice (Figure 2B).
Loss of host Fgl2 decreases infiltration of MDSCs in syngeneic transplanted lung tumors. Briefly, 5 x 105 LLC cells were subcutaneously injected in WT (n=10) or Fgl2-KO (n=9) mice. Tumors were harvested on day 15 after LLC cell implantation. Flow cytometric analysis of leukocyte subpopulations in tumor tissue from WT and Fgl2-KO mice: total leukocytes (CD45+, A), MDSCs (CD11b+Gr-1+, B), TAMs (F480+CD206+, C), DCs (CD11c+MHCII+, D) and Tregs (CD4+Foxp3+, E). Data are expressed as the percentage of positive cells (mean ± SEM). The results shown are representative of three independent experiments. *p<0.05, **p<0.01 (two-tailed unpaired Student's t-test) versus the indicated comparisons.
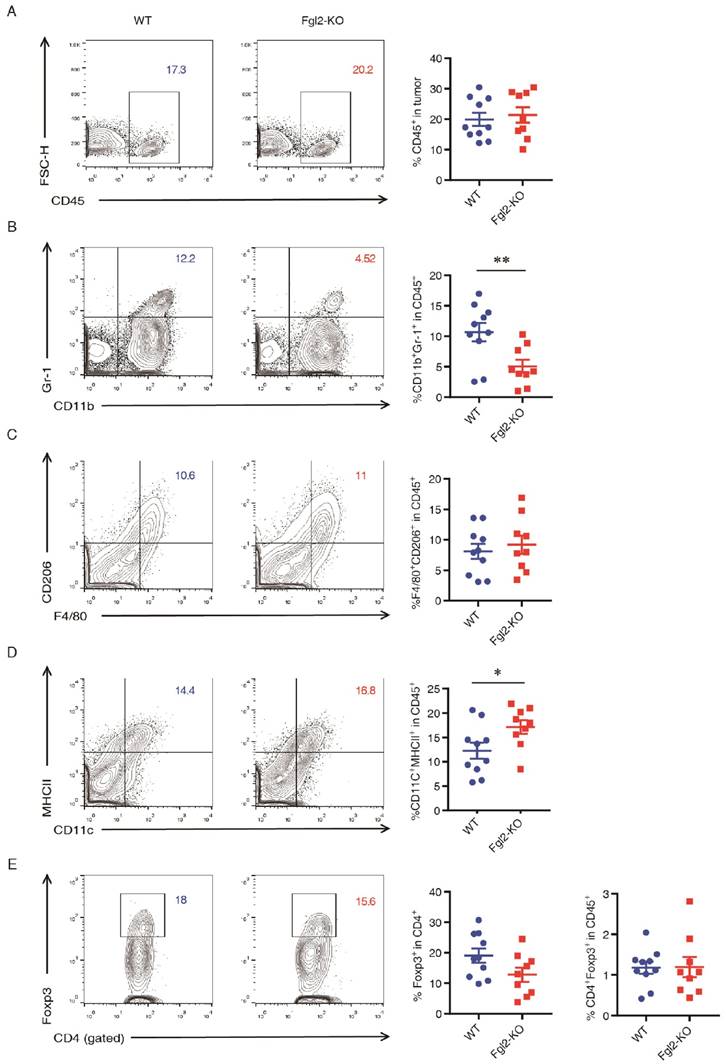
In addition, the proportions of TAMs (F4/80+CD206+) and Treg cells (CD4+Foxp3+) in WT and Fgl2-KO mice were also similar (Figure 2 C&E). As Fgl2 is known to suppress DC maturation by binding to FcγRIIB [8], we also noticed a slight increase in the frequency of mature DCs (CD11c+MHCII+) in the tumors of Fgl2-KO mice compared to those from WT mice (Figure 2D).
Fgl2 promotes CXCL12-mediated accumulation of MDSCs in the TME
As is knowniNOS (encoded by NOS2) and arginase (encoded by ARG1), which are enzymes involved in the metabolism of L-arginine, have been defined as functional markers for MDSCs [22, 23]. We detected the expression of NOS2 and ARG1 in the WT and Fgl2 tumors (Figure 3A&B). In addition, we found that the expression of NOS2 and ARG1 was significantly down-regulated in the tumors of Fgl2-KO mice (Figure 3A&B). Consistently, the effector cytokines of MDSCs including VEGF and TGF-β were decreased in Fgl2-KO tumors (Figure 3C&D).
Fgl2 promotes CXCL12-mediated accumulation of MDSCs in the TME. Implanted tumors were harvested from WT and Fgl2-KO mice bearing LLC tumors for 15 days. The expression of NOS2, Arg1, VEGF and TGF-β was measured in tumors of WT and Fgl2-KO mice by qRT-PCR (A-D). The frequency of MDSCs (CD11b+Gr1+) in the spleen and bone marrow was detected in naïve as well as in tumor-bearing WT and Fgl2-KO mice (E&F). The percentage of Ki67+ MDSCs (gated within CD11b+Gr-1+) in tumors of WT and Fgl2-KO mice was calculated by flow cytometry (G). The expression of CXCL12 and CXCL5 was detected in tumors of WT and Fgl2-KO mice by qRT-PCR (H-I). Analysis of the RNAseq expression data (n=178 NSCLC patients) from The Cancer Genome Atlas (TCGA). Pearson correlation analyses were utilized to examine the correlations between CXCL12, CD14 and CD33 (J&K). All of the data are presented as the mean ± SEM. *p<0.05, ***p<0.001 (two-tailed unpaired Student's t-test) versus the indicated comparisons.
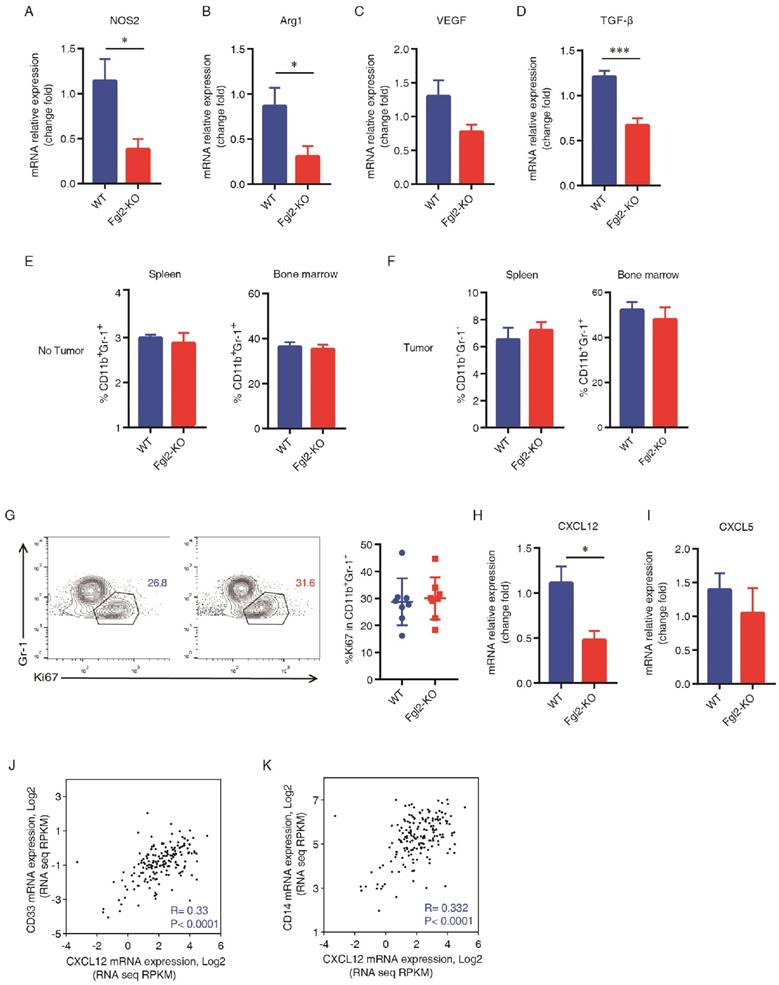
To further explore the role of Fgl2 in MDSC-induced tumor progression, we compared the percentage of MDSCs in the bone marrow and spleen of mice during tumor progression. We first confirmed that the frequency of MDSCs both in the bone marrow and spleen was similar at baseline in the two genetic backgrounds (Figure 3E). The proportion of MDSCs was increased in the spleen and bone marrow in response to LLC-tumor inoculation both in WT and Fgl2-KO mice (Figure 3E&F). However, the frequency of MDSCs in the spleen and bone marrow of WT and Fgl2-KO tumor-bearing mice was also similar (Figure 3F).
To further understand the mechanisms of the decreased MDSC accumulation in tumors of Fgl2-KO mice, we explored the effect of Fgl2 on the proliferation of MDSCs. A similar proliferation rate of MDSCs was found in tumors of WT and Fgl2-KO mice, as tested by Ki67 staining (Figure 3G).We then asked whether chemokines in the TME contributed to the reduction of MDSCs in tumors of Fgl2-KO mice. Previous studies have shown that CXCL12-CXCR4 and CXCL5-CXCR2 are the primary pathways mediating the recruitment of MDSCs into the TME [24, 25]. Therefore, we detected the expression of CXCL12 and CXCL5 in the tumors, and we found that CXCL12 was decreased in the tumors of Fgl2-KO mice compared with tumors of WT mice (Figure 3H&I). Further evidence that CXCL12 may affect the recruitment of MDSCs was obtained in human NSCLC using gene expression data from The Cancer Genome Atlas (TCGA) portal (http://www.cbioportal.org/ Accessed December 3, 2016). In line with the above mouse data, CXCL12 gene expression was indeed correlated with MDSC markers (CD33 and CD14) in the tumors of NSCLC patients (Figure 3J&K).
Fgl2 enhances the activation and function of CAFs directly
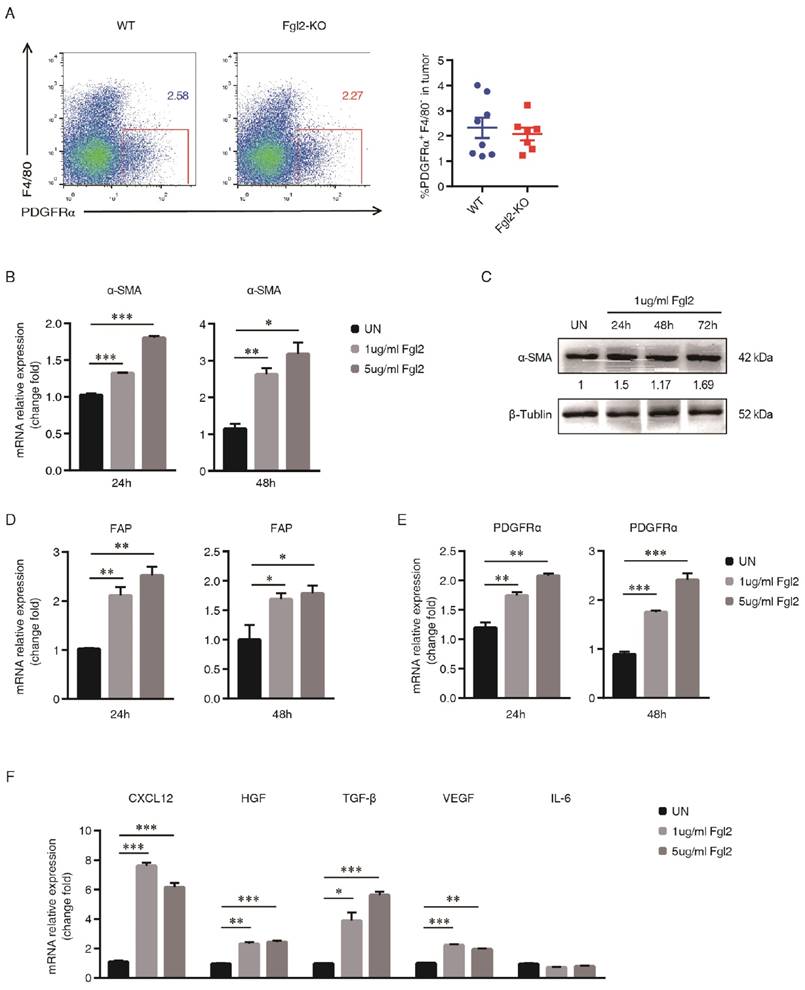
According to previous studies, CAFs are the principal source of CXCL12 in the TME. The TME comprises a complex milieu of various types of cells, and CAFs are the most abundant cells in the majority of carcinomas [26, 27]. CAFs are recruited to the TME supporting cancer cell proliferation, invasion and metastasis directly [28, 29]. More importantly, CAFs modulates the immune response to help malignant cells evade immune surveillance to promote tumor progression [30-32]. Considering the vast heterogeneity of CAFs, we chose PDGFRα as a CAF marker based on previous study [15]. PDGFRα is abundantly expressed by CAFs, including but not restricted to FAP+ and α-SMA+ CAFs [28]. We observed that the frequency of CAFs (PDGFRα+F4/80-) was not significantly different in the tumors of WT mice compared with that in Fgl2-KO mice (Figure 4A). To investigate whether the phenotype and function of CAFs could be directly regulated by Fgl2, CAFs were isolated from the LLC tumors implanted in C57BL/6 mice by FACS as described and incubated with recombined Fgl2 (1 µg/ml or 5 µg/ml). α-SMA, which usually identifies CAFs with myofibroblast morphology, enhances the activation phenotype of CAFs [33]. We found that both the mRNA and protein levels of α-SMA were up-regulated by recombiant Fgl2 stimulation (Figure 4B&C). In addition, FAP possessing dipeptidyl peptidase and collagenase activities is essential for degrading the extracellular matrix (ECM) and stimulating tumor growth; meanwhile, PDGF-PDGFRα has been shown important to initiate the desmoplastic reaction in CAFs [34]. Consistent with what we found previously, our data showed that Fgl2 directly enhanced FAP and PDGFRα expression in CAFs, further verifying that Fgl2 could promote the phenotype and function of CAFs (Figure 4D&E). We also found elevation of CXCL12 in the Fgl2-treated CAFs, indicating that Fgl2 could induce CXCL12-CXCR4 pathway activation in the TME (Figure 4F). Moreover, the levels of TGF-β, HGF and VEGF, which are highly secreted by CAFs [34], were elevated in the Fgl2-treated group compared with the NC group, but there was no difference in the expression of IL-6 between the two groups (Figure 4F). Taken together, Fgl2 directly enhanced the activation and function of CAFs in a murine tumor model.
Fgl2 induces an activated and pro-tumorigenic phenotype of CAFs. The percentage of CAFs (PDGFRα+F4/80- ) in tumors of WT and Fgl2-KO mice was calculated by flow cytometry 15 days after tumor injection (A). CAFs were obtained by sorting PDGFRα+F4/80- cells from tumors of C57BL/6 LLC tumor-bearing mice, and treated with recombinant mouse Fgl2 (1 µg/ml or 5 µg/ml) for 24, 48 and 72 hours. The expression of CAF activation-associated genes (α-SMA, B&C; FAP, D; and PDGFRα, E) was evaluated by qRT-PCR and western blotting. The signal intensities of the western blot were quantified using software ImageJ (version 1.47), and the ratio of α-SMA/β-tubulin is presented. The CAF-secreted cytokines (CXCL12, HGF, TGF-β, VEGF and IL-6) were measured by qRT-PCR at 48 hours post Fgl2 stimulation (F). All data represent one out of three independent experiments and are presented as the mean ± SEM. *p<0.05, **p<0.01, ***p<0.001 (A, two-tailed unpaired Student's t-test; B & D-F, ANOVA followed by Dunnett's test for multiple comparisons) versus the indicated comparisons.
Expression of Fgl2 in human NSCLC is correlated with CAF activation and function
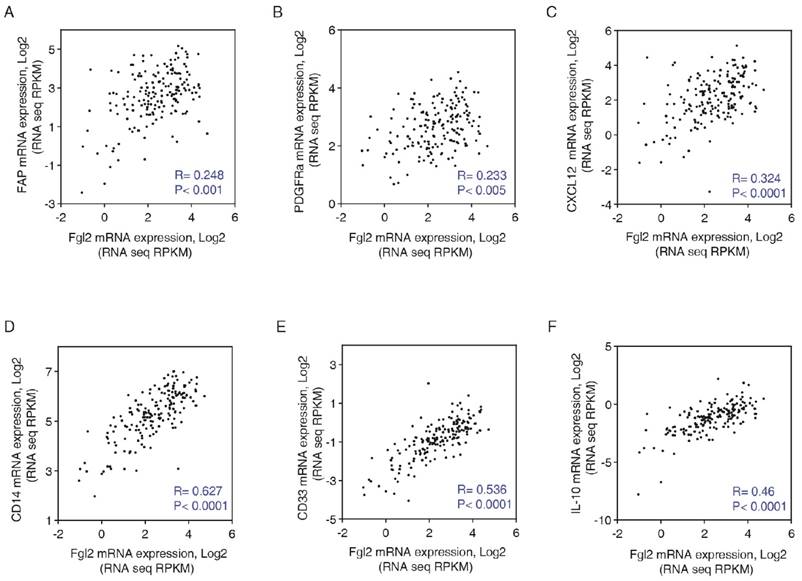
The results till now proven that stroma-derived Fgl2 promote CAFs activating and MDSCs accumulation. To validate the observed correlations from mouse tumor model, we assessed the correlations in human NSCLC. Using RNAseq expression data (n=178 NSCLC patients) from The Cancer Genome Atlas (TCGA), Pearson correlation analyses were utilized to examine the correlations between Fgl2 expression and CAFs. Consistent with our findings, Fgl2 expression was positively correlated with the expression of with CAF markers and effectors, such as FAP, PDGFRα, CXCL12. Furthermore, MDSC markers and effectors, including CD33, CD14 and IL-10 were found to be positively correlated with the expression of Fgl2 (Figure 5). These data suggest that augmented Fgl2 expression in human NSCLC is correlated with enhanced activation of CAFs and elevated levels of CAF-derived chemokine CXCL12, which results in the recruitment of MDSCs.
Expression of Fgl2 in human NSCLC is correlated with CAF activation and function. Pearson's test was used to analyze the correlation between gene expression of Fgl2 and CAF markers and effectors (FAP, A; PDGFRα, B; and CXCL12, C) and MDSC markers and effectors (CD14, D; CD33, E; and IL-10, F). mRNA expression data for these genes were downloaded from TCGA (n=178, NSCLC patients). R = Pearson correlation coefficient.
In conclusion, our data indicate a critical functional role of stroma-derived Fgl2 in promoting CAF activation and tumor-promoting functions in lung cancer.
Discussion
CAFs, the most abundant “non-cancerous” cells, promote the tumor procession by encouraging tumor growth, angiogenesis and metastasis [35]. However, the mechanisms involved in CAF activation and function are still largely unknown. Herein, we explored a previously unrecognized role of stroma-derived Fgl2 in promoting CAF activation and tumor-promoting functions in lung cancer.
In the present study, we have proven that deficiency of host Fgl2 is associated with reduced growth of transplanted LLC tumors. In addition, the tumor-promoting effects of Fgl2 are mediated via the host immune system, with decreases in the percentage of tumor-infiltrating MDSCs in the Fgl2-KO mice. The impacts of Fgl2 on tumorigenesis and metastasis have primarily been investigated in immune-deficient mice, and the studies focused on the functional changes in tumor cells that were intervened Fgl2 expression [10-12]. However, multiple studies have demonstrated that Fgl2 plays an important role in the pathogenesis of inflammatory disorders by regulating both innate and adaptive immune responses [7, 8]. Interestingly, new research regarding Fgl2 in GBM has shown that overexpression of Fgl2 in glioma cells does not affect the proliferation of tumor cells directly, but functions as an immune-suppressive modulator in the anti-tumor immune response, including by enhancing MDSCs accumulation [13]. Nevertheless, several previous studies have determined that immune cells and stromal cells express Fgl2 during inflammatory disorders [36, 37]. The above studies are consistent with our findings that tumor stroma-derived Fgl2 plays an essential role in tumor progression.
Our study also identifies that tumor stroma-derived Fgl2 induces an activated, pro-tumorigenic CAF phenotype in lung carcinoma. The TME comprises various types of cells, and CAFs are the most abundant cells in the majority of carcinomas [26, 27]. CAFs are recruited to the TME supporting tumor cell proliferation, invasion and metastasis directly [28, 29]. More importantly, CAFs modulate the immune response to help malignant cells evade immune surveillance to support tumor progression [30-32]. To date, numerous studies have indicated that the plasticity of CAFs may provide new mechanisms and therapeutic strategies in tumors [34]. We have found that Fgl2 up-regulated the expression of α-SMA, FAP and PDGFRα, which demonstrated an enhanced activation phenotype of CAFs. As is known, CAFs are identified with myofibroblast morphology and phenotype [38]. Interestingly, a previous study in chronic HCV infection found that the level of Fgl2 in the patient plasma was correlated with the severity of liver fibrosis which is closely related to the activation of myofibroblasts [39]. This observation is in accordance with our study, suggesting that Fgl2 is an important molecule in fibroblast activation, both in fibroblast-associated tumor progression and in fibroblast-associated liver fibrosis.
CAFs function principally through secretion of growth factors, chemokines and cytokines [38, 40, 41]. In our study, we confirmed that Fgl2 promotes CXCL12 expression in CAFs, which was reported to be involved in boosting the proliferation of carcinoma cells, stimulating neoangiogenesis and the recruitment of MDSCs [42-44]. In return, CXCL12 might enable CAFs to maintain a tumor myofibroblastic phenotype in an autocrine manner, as has been previously reported. In addition, a previous study in pancreatic cancer reveals that FAP+ CAFs are the principal source of CXCL12, and administering AMD3100, an inhibitor of CXCR4 which is a CXCL12 receptor, synergized with α-PD-L1 and α-CTLA-4 by enhancing the anti-tumor immune response [45]. Furthermore, our current results reveal that Fgl2 also up-regulated TGF-β, HGF and VEGF in CAFs, which have been identified as CAF pro-tumorigenic effectors, contributing to the establishment of an immunosuppressive TME and drug resistance as well as neoangiogenesis [34]. Our results implicate Fgl2 in functional activation of CAFs, contributing to the formation of pro-tumorigenic stroma.
Taken together, our findings highlight a novel role of stroma-derived Fgl2 in CAF activation and function, as well as suggest a fibroblast-dependent mechanism for Fgl2-mediated tumor progression. The exact mechanisms underlying this phenomenon warrant further study.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Dr. Steve Smiley (The Trudeau Institute, NY, USA) for kindly providing Fgl2-knockout mice. We would also like to thank Xiaolan Fu (Third Military Medical University) for technical assistance with flow-cytometry.
Funding
This work was supported by the National Nature Science Foundation of China (No. 81472648 and No. 81620108023 to Bo Zhu, No. 31570866, No. 31170838 to Bo Guo).
Author contributions
Conception and design: Bo Zhu, Bo Guo and Ying Zhu.
Development of methodology: Ying Zhu and Longhui Zhang
Acquisition of data (provided animals acquired and, provided facilities, etc.): Ying Zhu, Longhui Zhang, Haoran Zha, Fei Yang and Chunyan Hu
Analysis and interpretation of data (e.g., statistical analysis, biostatistics and computational analysis): Ying Zhu and Longhui Zhang
Writing, review, and/or revision of the manuscript: Bo Zhu, Bo Guo and Ying Zhu
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): Ying Zhu, Longhui Zhang and Haoran Zha
Competing Interests
The authors have declared that no competing interest exists.
References
1. Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nature reviews Immunology. 2015;15:669-82
2. Brock A, Krause S, Ingber DE. Control of cancer formation by intrinsic genetic noise and microenvironmental cues. Nature reviews Cancer. 2015;15:499-509
3. Liu J, Tan Y, Zhang J. et al. C5aR, TNF-alpha, and FGL2 contribute to coagulation and complement activation in virus-induced fulminant hepatitis. Journal of hepatology. 2015;62:354-62
4. Liu Y, Xu S, Xiao F. et al. The FGL2/fibroleukin prothrombinase is involved in alveolar macrophage activation in COPD through the MAPK pathway. Biochemical and biophysical research communications. 2010;396:555-61
5. Clark DA, Foerster K, Fung L. et al. The fgl2 prothrombinase/fibroleukin gene is required for lipopolysaccharide-triggered abortions and for normal mouse reproduction. Molecular human reproduction. 2004;10:99-108
6. Ghanekar A, Mendicino M, Liu H. et al. Endothelial induction of fgl2 contributes to thrombosis during acute vascular xenograft rejection. Journal of immunology. 2004;172:5693-701
7. Joller N, Lozano E, Burkett PR. et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. 2014;40:569-81
8. Chan CW, Kay LS, Khadaroo RG. et al. Soluble fibrinogen-like protein 2/fibroleukin exhibits immunosuppressive properties: suppressing T cell proliferation and inhibiting maturation of bone marrow-derived dendritic cells. Journal of immunology. 2003;170:4036-44
9. Liu H, Shalev I, Manuel J. et al. The FGL2-FcgammaRIIB pathway: a novel mechanism leading to immunosuppression. European journal of immunology. 2008;38:3114-26
10. Yuwaraj S, Ding J, Liu M. et al. Genomic characterization, localization, and functional expression of FGL2, the human gene encoding fibroleukin: a novel human procoagulant. Genomics. 2001;71:330-8
11. Su K, Chen F, Yan WM. et al. Fibrinogen-like protein 2/fibroleukin prothrombinase contributes to tumor hypercoagulability via IL-2 and IFN-gamma. World journal of gastroenterology. 2008;14:5980-9
12. Liu Y, Xu L, Zeng Q. et al. Downregulation of FGL2/prothrombinase delays HCCLM6 xenograft tumour growth and decreases tumour angiogenesis. Liver international: official journal of the International Association for the Study of the Liver. 2012;32:1585-95
13. Yan J, Kong LY, Hu J. et al. FGL2 as a Multimodality Regulator of Tumor-Mediated Immune Suppression and Therapeutic Target in Gliomas. Journal of the National Cancer Institute. 2015:107
14. Hu J, Yan J, Rao G. et al. The Duality of Fgl2 - Secreted Immune Checkpoint Regulator Versus Membrane-Associated Procoagulant: Therapeutic Potential and Implications. International reviews of immunology. 2014
15. Sharon Y, Alon L, Glanz S. et al. Isolation of normal and cancer-associated fibroblasts from fresh tissues by Fluorescence Activated Cell Sorting (FACS). Journal of visualized experiments: JoVE. 2013 e4425
16. Gao J, Aksoy BA, Dogrusoz U. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1
17. Cerami E, Gao J, Dogrusoz U. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2:401-4
18. Qin WZ, Li QL, Chen WF. et al. Overexpression of fibrinogen-like protein 2 induces epithelial-to-mesenchymal transition and promotes tumor progression in colorectal carcinoma. Medical oncology. 2014;31:181
19. Woo EY, Chu CS, Goletz TJ. et al. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer research. 2001;61:4766-72
20. Sawant A, Schafer CC, Jin TH. et al. Enhancement of antitumor immunity in lung cancer by targeting myeloid-derived suppressor cell pathways. Cancer research. 2013;73:6609-20
21. Dai F, Liu L, Che G. et al. The number and microlocalization of tumor-associated immune cells are associated with patient's survival time in non-small cell lung cancer. BMC cancer. 2010;10:220
22. Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nature reviews Immunology. 2005;5:641-54
23. Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunological reviews. 2008;222:180-91
24. Yang L, Huang J, Ren X. et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer cell. 2008;13:23-35
25. Liu Y, Lai L, Chen Q. et al. MicroRNA-494 is required for the accumulation and functions of tumor-expanded myeloid-derived suppressor cells via targeting of PTEN. Journal of immunology. 2012;188:5500-10
26. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
27. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer cell. 2012;21:309-22
28. Erez N, Truitt M, Olson P. et al. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer cell. 2010;17:135-47
29. Olumi AF, Grossfeld GD, Hayward SW. et al. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer research. 1999;59:5002-11
30. Singh S, Ross SR, Acena M. et al. Stroma is critical for preventing or permitting immunological destruction of antigenic cancer cells. The Journal of experimental medicine. 1992;175:139-46
31. Iijima J, Konno K, Itano N. Inflammatory alterations of the extracellular matrix in the tumor microenvironment. Cancers. 2011;3:3189-205
32. Liao D, Luo Y, Markowitz D. et al. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PloS one. 2009;4:e7965
33. Polanska UM, Orimo A. Carcinoma-associated fibroblasts: non-neoplastic tumour-promoting mesenchymal cells. Journal of cellular physiology. 2013;228:1651-7
34. Ohlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. The Journal of experimental medicine. 2014;211:1503-23
35. Madar S, Goldstein I, Rotter V. 'Cancer associated fibroblasts'-more than meets the eye. Trends in molecular medicine. 2013;19:447-53
36. Shalev I, Wong KM, Foerster K. et al. The novel CD4+CD25+ regulatory T cell effector molecule fibrinogen-like protein 2 contributes to the outcome of murine fulminant viral hepatitis. Hepatology. 2009;49:387-97
37. Sun Y, Xi D, Ding W. et al. Soluble FGL2, a novel effector molecule of activated hepatic stellate cells, regulates T-cell function in cirrhotic patients with hepatocellular carcinoma. Hepatology international. 2014;8:567-75
38. Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes & development. 2016;30:1002-19
39. Foerster K, Helmy A, Zhu Y. et al. The novel immunoregulatory molecule FGL2: a potential biomarker for severity of chronic hepatitis C virus infection. Journal of hepatology. 2010;53:608-15
40. Pickup M, Novitskiy S, Moses HL. The roles of TGFbeta in the tumour microenvironment. Nature reviews Cancer. 2013;13:788-99
41. Moskovits N, Kalinkovich A, Bar J. et al. p53 Attenuates cancer cell migration and invasion through repression of SDF-1/CXCL12 expression in stromal fibroblasts. Cancer research. 2006;66:10671-6
42. Obermajer N, Muthuswamy R, Odunsi K. et al. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer research. 2011;71:7463-70
43. Domanska UM, Timmer-Bosscha H, Nagengast WB. et al. CXCR4 inhibition with AMD3100 sensitizes prostate cancer to docetaxel chemotherapy. Neoplasia. 2012;14:709-18
44. Li X, Ma Q, Xu Q. et al. SDF-1/CXCR4 signaling induces pancreatic cancer cell invasion and epithelial-mesenchymal transition in vitro through non-canonical activation of Hedgehog pathway. Cancer letters. 2012;322:169-76
45. Feig C, Jones JO, Kraman M. et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:20212-7
Author contact
![]() Corresponding authors: Bo Guo (PhD), Department of Pathogenic Biology, Third Military Medical University, Chongqing 400037, China; E-mail: guoboedu.cn; Tel (Fax): 86-23-68755646. Bo Zhu (PhD & MD), Institute of Cancer, Xinqiao Hospital, Third Military Medical University, Chongqing 400037, P.R. China; E-mail: bo.zhuedu.cn; Tel (Fax): 86-23-68755626.
Corresponding authors: Bo Guo (PhD), Department of Pathogenic Biology, Third Military Medical University, Chongqing 400037, China; E-mail: guoboedu.cn; Tel (Fax): 86-23-68755646. Bo Zhu (PhD & MD), Institute of Cancer, Xinqiao Hospital, Third Military Medical University, Chongqing 400037, P.R. China; E-mail: bo.zhuedu.cn; Tel (Fax): 86-23-68755626.