Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(10):1297-1308. doi:10.7150/ijbs.21172 This issue Cite
Research Paper
Diabetes Induces Abnormal Ovarian Function via Triggering Apoptosis of Granulosa Cells and Suppressing Ovarian Angiogenesis
Yanqing Wu1* ![]() , Yiyang Li2*, Xinghui Liao3*, Zhengchao Wang4, Rui Li2, Shuang Zou2, Ting Jiang2, Bingbing Zheng5, Ping Duan6, Jian Xiao2
, Yiyang Li2*, Xinghui Liao3*, Zhengchao Wang4, Rui Li2, Shuang Zou2, Ting Jiang2, Bingbing Zheng5, Ping Duan6, Jian Xiao2 ![]()
1. The Institute of Life Sciences, Wenzhou University, Wenzhou, Zhejiang 325035, China;
2. Molecular Pharmacology Research Center, School of Pharmaceutical Science, Wenzhou Medical University, Wenzhou, Zhejiang 325035, China;
3. Department of Molecular Pathology Laboratory, Zhejiang Rongjun Hospital, Jiaxing Zhejiang 314000;
4. College of Life Sciences, Fujian Normal University,Fuzhou 350007, China;
5. Department of Obstetrics, First Affiliated Hospital of Wenzhou Medical University, Zhejiang, China;
6. Department of Obstetrics and Gynecology, The Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University, Wenzhou 325027, China.
* These authors contributed equally to this work.
Received 2017-5-24; Accepted 2017-7-20; Published 2017-10-17
Abstract

Diabetes triggers abnormal ovarian follicular development and consequently leads to infertility. Here, we established a type 2 diabetes mouse model by feeding with high fat diet (HFD) for 15/20 weeks and assessed the effect of diabetes on follicular development and ovarian angiogenesis. After fed with HFD for 15 weeks, mice had the characteristics of type 2 diabetes, which was much more serious after 20 weeks on HFD. After 20 weeks on HFD, the mice had shown abnormal ovarian morphology with hyaline appearance, much less blood vessel, follicular development arrest and less of granulosa cells (GCs) in mature follicles, but not in ovaries from 15 weeks on HFD. Elevated makers of DNA damage, ER stress and apoptosis of GCs were observed in ovaries from HFD for 20 weeks. Additionally, diabetes significantly suppressed ovarian angiogenesis with the evidence of down-regulation of CD31 via inhibiting HIF1α-VEGF signaling pathway in time-dependent. We concluded that diabetes triggers abnormal ovarian function via inducing GCs apoptosis and suppressing ovarian angiogenesis.
Keywords: diabetes, granulosa cells (GCs), apoptosis, angiogenesis, DNA damage.
Introduction
Mammalian ovarian follicles develop from primordial follicles to antral follicles, which is essential for normal reproductive function of female. Increasing prevalence of obesity, type 2 diabetes, and related complications are major health issues in world. Evidence is growing that chronic hyperglycemia has a detrimental effect on follicular development and consequently leads to infertility [1-4]. But the exact mechanisms that responsible for pathogenesis of diabetes-induced infertility are not clear.
The growing mammalian follicle consists of a single oocyte, surrounding granulosa cells (GCs), and an outer rim of theca cells. The growth of oocytes and GCs development are mutually regulatory events during follicular development. GCs not only provide various energy substrates for nurturing oocytes, but also have an essential role on corpus luteum (CL) formation, which influences the quality of oocytes and subsequent embryonic development. The apoptosis of GCs is the main molecular mechanism of atresia [5, 6]. Our and others' prior researches have delineated that obesity and diabetes significantly induced the apoptosis of GCs [3, 7]. But the exact mechanisms are still unclear.
Our and others' studies have revealed that elevated oxidative stress, endoplasmic reticulum (ER) stress, and consequent excessive cell apoptosis are the causal events for diabetes-related complications [8-10]. Diabetes significantly triggers oxidative stress, which in turn leads to DNA damage [11]. DNA damage triggers cell cycle arrest and results in apoptosis, which suppresses oocytes maturation [12]. Thus, we propose that diabetes significantly induced apoptosis of GCs by triggering DNA damage and ER stress during ovarian follicular development and CL formation, consequently leading to abnormal follicular development.
Prior studies have demonstrated that the vascular system in ovary originates from theca folliculi interna and breaks up into a rich network of capillaries that builds a basket-like network around the avascular stratum granulosum [13, 14]. The movement of nutrients and small macromolecules from blood vessels of theca into follicular fluid is essential for the development of the ovum and follicle. Angiogenesis is required throughout all processes of follicular development, including ovulatory cycle and CL formation [15, 16]. Recent studies have reported that vascular endothelial growth factor (VEGF) has a critical role in the process of thecal angiogenesis during follicular development [17, 18]. As one of crucial transcriptional factor, hypoxia inducible factor 1α (HIF1α) participates in the regulation of VEGF expression in ovary [19-21]. Chronic exposure to hyperglycemia significantly suppresses VEGF expression and inhibits angiogenesis [22].
As the rapidly increasing number of women with type 2 diabetes, it is an urgent need to establish a useful and effective diabetic animal model to reveal the effect of diabetes on ovarian development. In present study, 4-week old female mice were fed with high fat diet (HFD) to establish type 2 diabetes model and used to reveal the molecular mechanisms underlying diabetes on ovarian follicular development, offering a novel theoretical basis for diabetes related-infertility.
Material and Methods
Induction of Type 2 Diabetes Mouse Model
4-week old female C57BL/6J mice were purchased from the Animal Center of the Chinese Academy of Sciences in Shanghai, China. The protocol for animal care and use was conformed to the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health and was approved by the Animal Care and Use Committee of Wenzhou University. The animals were maintained under a 14-h light/10-h dark condition. After arrived, mice were divided into two groups and fed either with a high-fat diet (HFD, Research Diets, New Brunswick, NJ) or a normal control diet (NC, Research Diets, Harlan, US) for 15 weeks or 20 weeks. Body weight was recorded weekly. After 15/20 weeks fed with HFD, body mass index (BMI), blood glucose levels, glucose tolerance test (GTT) and insulin tolerance test (ITT) were assessed after fed 15 weeks on HFD and 20 weeks on HFD.
Blood Glucose Measurement, Glucose Tolerance Test and Insulin Tolerance Test
Blood glucose was measured using a handheld glucometer with appropriate test strips (FreeStyle Lite). For GTT, mice were fasted overnight (17:00-9:00) and then intraperitoneally injected with glucose at dose of 2 g/kg body weight. Blood glucose levels were measured before injection and at 15, 30, 60, 90, and 120 min after injection. For ITT, mice were fasted for 5 h (9:00-14:00) and then intraperitoneally injected with insulin at dose of 0.75 units/kg body weight. Blood glucose levels were measured before injection and at 15, 30, 60, 90, and 120 min after injection.
Hematoxylin and Eosin (H&E) and Immunohistochemistry
Ovaries were collected at 15 weeks or 20 weeks and fixed with 4% paraformaldehyde in phosphate buffered saline (PBS). Ovaries were dehydrated in alcohol and embedded with paraffin. 7 µm sections were dewaxed and hydrated, then stained with H&E and eventually observed under light microscope. For immunohistochemistry, the sections were also incubated in 3% H2O2 for 15 min and then in blocking solution for 45min after dewaxing and hydration. Subsequently, the sections were incubated at 4°C overnight with the following primary antibodies: anti-CD31 (1:250, sc793); anti-VEGF (1:500, sc507) and anti- C/EBP-homologous protein (CHOP) (1:250, sc10790). After washed with PBS for 3 times, the sections were incubated with horseradish peroxidase-conjugated secondary antibodies for 4 h at 37°C. The sections were reacted with 3, 3-diaminobenzidine (DAB). The results were imaged using a Nikon A1 Plus fluorescence microscope (Nikon, Tokyo, Japan).
Lipid Hydroperoxide Quantification (LPO)
The degree of lipid peroxidation, an index of oxidative stress, was quantitatively assessed using the Calbiochem Lipid Hydroperoxide Assay Kit (Milliproe, Bedford, MA). Briefly, ovaries were homogenized in HPLC-grade water. Lipid hydroperoxides were extracted from ovary by deoxygenated chloroform, and the absorbance at 500 nm was measured after reaction with chromogen. Results were expressed as μM lipid hydroperoxides per microgram protein. Protein concentrations were determined with BioRad DC protein assay kit (BioRad, Hercules, CA).
Immunoblotting
The ovaries from different experimental groups were sonicated in lysis buffer, containing a protease inhibitor cocktail (Sigma, St Louis, MO). Protein concentrations were determined using the bicinchoninic acid (BCA) method (Thermo, Rockford, IL, USA). Equal amounts of protein and the Precision Plus Protein Standards (Bio-Rad) were resolved by SDS-PAGE electrophoresis and transferred onto PVDF membranes (Bio-Rad). Membranes were incubated with 5% nonfat milk for 45 min and then incubated for 18 h at 4℃ with the primary antibodies at dilution of 1:1,000 in 5% nonfat milk. To test whether equivalent amounts of protein were loaded among all samples, membranes were stripped and incubated with a mouse antibody against β-actin (Abcam) to generate a signal used as a loading control. Signals were visualized using the ChemiDocTM XRS+ Imaging System (Bio-Rad), and band densities were quantified with Multi Gauge Software for Science Lab 2006 (Fuji Film Corporation, Japan). The quantitative analysis of protein was calculated as the densitometric value of phosphorylated protein level/the densitometric value of unphosphorylated protein. All experiments were repeated in triplicate with the use of independently prepared tissue.
Real-time PCR
Using the TRIzol reagent, mRNA was isolated from each ovary, and then reversed transcribed using the high-capacity cDNA archive kit (Applied Biosystem, Grand Island, NY). RT-PCR for HIF1a, VEGF and β-actin were performed using the Maxima SYBR Green/ROX qPCR Master Mix assay (Thermo Scientific, Rockford, IL) in the StepOne Plus system (Applied Biosystem).
TUNEL Assay
TUNEL staining (ApopTag Fluorescein in Situ Apoptosis Detection kit, Chemicon) was performed to determine apoptosis level. After dewaxing and hydration, ovarian sections were incubated with 20 μg/ml proteinase K working solution for 15 min at 37°C. The slides were rinsed with PBS for 3 time followed by incubation with TUNEL reaction mixture for 1 h at 37℃. After rinsed with PBS (5 min, thrice), sections were incubated with 4', 6-diamidino-2-pheny-lindole (DAPI, Beyotime, Shanghai, China) for 5 min at room temperature and mounted with aqueous mounting medium(Sigma, St Louris, MO). The results were imaged using a Nikon A1 Plus fluorescence microscope (Nikon, Tokyo, Japan).
Statistical Analyses
Data were presented as means ± SE. Experiments were repeated at least three times, and ovarian sample from each replicate was from different female mice. Statistical differences were determined by one-way analysis of variance (ANOVA) using Graphpad Prism5. Tukey test was used to estimate the significance of the results.
Results
Diet induced-obese (DIO) type 2 diabetes caused abnormal ovarian follicular development
We used 4-week-old female mice to induce type 2 diabetes via fed with HFD. After fed with HFD for 15/20weeks, mean body weights and BMI of mice in HFD group were both significantly greater than those in mice from normal control groups (Figure 1A and B), indicating that the mice had been successfully induced into obese mouse model after 15 weeks on HFD. Additionally, blood glucose levels of mice in HFD groups were both significantly enhanced when compared with NC group, more importantly, mean blood glucose level of mice in 20W-HFD group was also significantly higher than that in mice from 15W-HFD group (Figure 1G). Moreover, GTT and ITT results had demonstrated that HFD significantly impaired glucose tolerance and induced insulin resistance of mice after 15 weeks and hereafter (Figure 1C, D, E and F).
Diet induced-obese (DIO) type 2 diabetes caused abnormal ovarian follicular development. (A) Body weight of mice during fed with different diet (n=8); (B) Body mass index (BMI) of mice after 15/20 weeks fed with normal diet and HFD (n=8); Blood glucose level during GTT after 15 weeks(C) and 20 weeks(D) fed with different diet(n=6); Blood glucose level during ITT after 15 weeks(E) and 20 weeks(F) fed with different diet (n=6). (G) Blood glucose level of mice after 15/20 weeks fed with normal diet and HFD (n=8); (H-O) Representative images of H&E staining shown morphology of ovaries from mice after 15/20 weeks fed with different diet, in H, I, J, K and M, scale bars= 150 μm; in L and N, scale bars = 15 μm; in O, scale bars = 30 μm. *P<0.05 vs. 15W-NC group; **P<0.01 vs. 15W-NC group; ***P<0.001 vs. 15W-NC group; #P<0.05 vs. 20 W-NC group; ## P<0.01 vs. 20 W-NC group; ### P<0.001 vs. 20 W-NC group. ***P<0.001 vs. 15W-NC group; ### P<0.001 vs. 20 W-NC group; … P<0.001 vs. 15W-HFD group. NC: normal control diet group; HFD: high fat diet group; CL: corpus luteum; OC: oocyte; GC: granulosa cell; W: weeks.
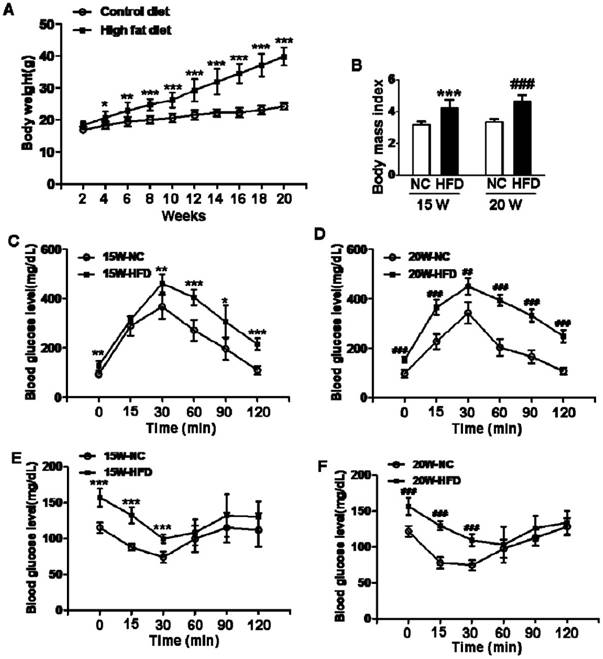
We assessed the effect of diabetes on follicular development. It was observed that the ovaries in mice had shown normal ovarian morphology with different stage of follicles and normal CL formation after fed with different diet for 15 weeks (Figure 1H and I). However, the majority of follicles in ovaries from 20W-HFD group were arrested at early stage or atretic (Figure 1M). Even some follicles had escaped and developed into matured follicles, the number of GCs was much less in cumulus-oocyte complex (COC) or follicular wall (Figure 1K, L, M, N and O), but no remarkable abnormal morphology in ovaries from mice fed with normal diet for 20 weeks (Figure 1J). Taken together, type 2 diabetes significantly induced apoptosis of GCs and arrested follicular development.
DIO-type 2 diabetes triggered oxidative stress and DNA damage of ovary
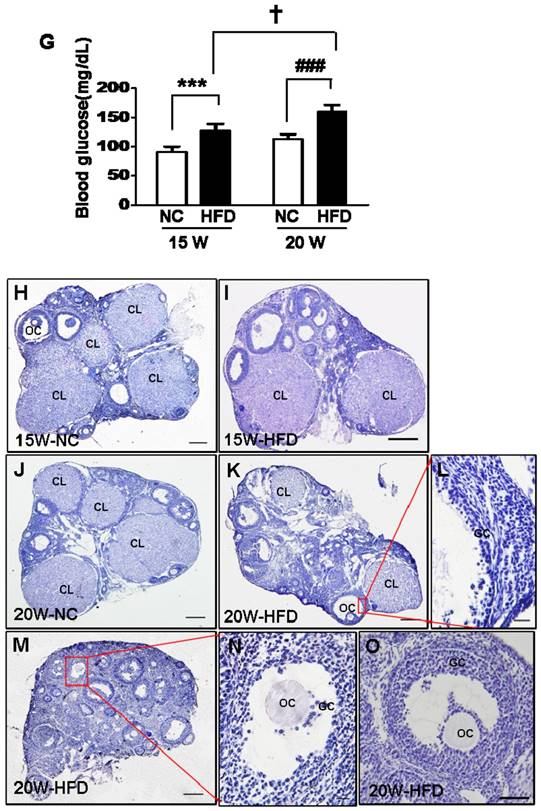
In current study, we had assessed whether oxidative stress and DNA damage were involved in type 2 diabetes-induced abnormal ovarian follicular development. We found that lipid peroxidation levels in ovaries from mice fed with HFD for 20 weeks were significantly higher than those in ovaries from the other groups (Figure 2A). The expressions of P53, p-H2AX and p-CHK2 in ovaries from 15/20 weeks HFD group were significantly elevated when compared with those in ovaries from NC group (Figure 2B, D, E and G). However, the expressions of p-CHK1 and p-cdc25 were significantly up-regulated in ovaries after 20 weeks fed with HFD, but no significant difference in ovaries from mice fed with different diet for 15 weeks (Figure 2B, C and F). Based on above data, it implied that the effect of DIO-type 2 diabetes on abnormal follicular development was time-dependent. Thus, the ovaries from mice after 20 weeks on different diet were used to further analyze the relevant mechanism hereafter.
DIO-type 2 diabetes triggered oxidative stress and DNA damages of ovary. (A) LPO levels in ovaries. (B) The protein levels of p-H2AX, p-CHK1, p-CHK2, p-cdc25 and P53 in ovaries from mice after 15/20 weeks fed with different diet. (C-G) Intensities of p-H2AX, p-CHK1, p-CHK2, p-cdc25 and P53 normalized to β-actin. Data are presented as the mean ± standard deviation (n=3); *P<0.05 vs. 15W-NC group; #P<0.05 vs. 20W-NC group. p‑:phosphorylated; NC: normal control group; HFD: high‑fat diet group; W: weeks.
DIO-type 2 diabetes induced ER stress of ovary
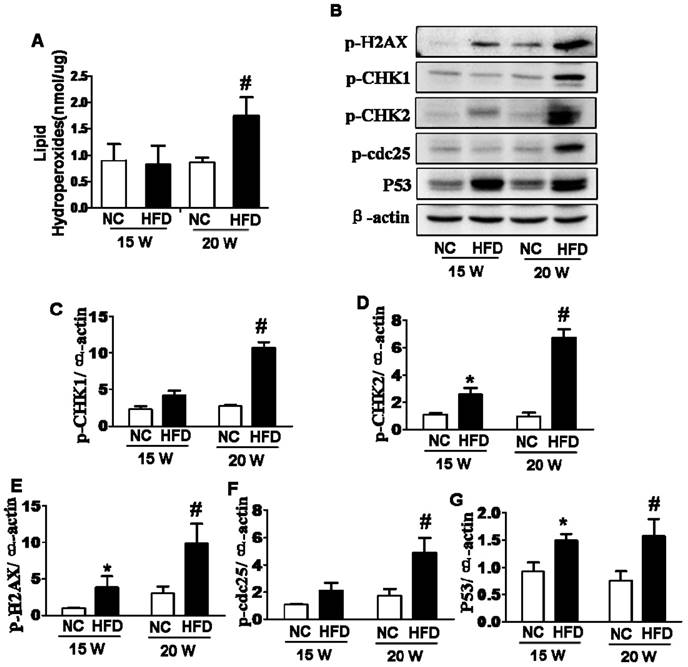
ER stress is the main molecular mechanism underlying diabetes-related complications [8, 10, 23, 24]. Thus, we detected the expressions of ER stress makers in ovaries and found that the expression levels of phosphorylated protein kinase RNA-like ER kinase (p-PERK), phosphorylated inositol requiring protein-1α (p-IRE1α), phosphorylated eukaryotic initiation factor 2α (p-eIF2α), CHOP and binding immunoglobulin protein (BiP) in ovaries from 20W-HFD group were significantly increased when compared with those in ovaries from 20W-NC group (Figure 3A and B). Immunohistochemical staining result was consistent with Western blot results with elevated expression of CHOP in ovaries from HFD group (Figure 3C).
DIO-type 2 diabetes induced ER stress of ovary. (A) The protein levels of p-PERK, p-IRE1α, p-eIF2α, CHOP and BiP in ovaries from mice after fed with different diet for 20 weeks. (B) Intensities of p-PERK normalized to PERK, p-IRE1α normalized to IRE1α, p-eIF2α normalized to eIF2α, CHOP and BiP normalized to β-actin. (C) Immunohistochemical staining of CHOP in ovaries from mice after fed with different diet for 20 weeks, scale bars= 30 μm. Data are presented as the mean ± standard deviation (n=3). #P<0.05 vs. 20W-NC group. NC: normal control diet group; HFD: high fat diet group; CL: corpus luteum; OC: oocyte; GC: granulosa cell; W: weeks.
DIO-type 2 diabetes led to caspase activation and subsequent apoptosis in ovary
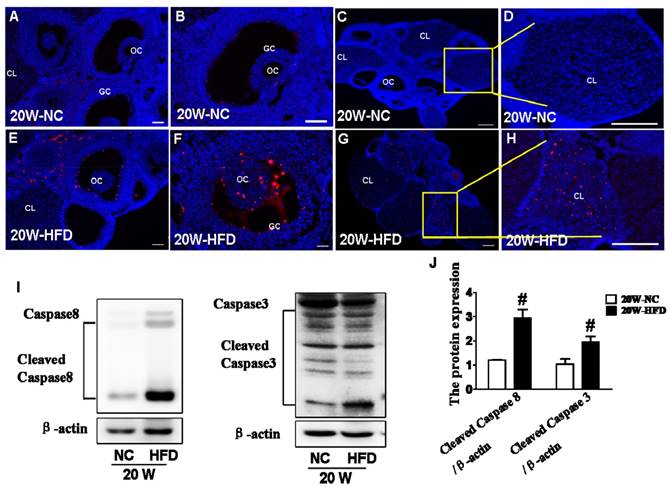
Here, we tried to further verify whether apoptosis was involved in the effect of DIO-type 2 diabetes on follicular development. TUNEL staining was used to detect the apoptotic cell in ovary. It was clearly observed that the apoptotic GCs surrounding oocytes and CL in ovaries from HFD group (Figure 4E, F, G and H) were much more than those in ovaries from NC group (Figure 4A, B, C and D). Moreover, expressions of cleaved-Caspase 8 and cleaved-Caspase 3 were significantly up-regulated in ovaries from HFD group (Figure 4I and J), indicating that DIO-type 2 diabetes significantly triggered apoptosis of GCs.
DIO-type 2 diabetes triggered caspase activation and subsequent apoptosis in ovary. (A) Representative images of TUNEL staining shown apoptotic cells (red signal) in ovaries from mice after fed with different diet for 20 weeks, in A and E, scale bars= 30 μm; in B, D, F and H, scale bars= 15 μm; in C and G, scale bars= 150 μm. (B) The protein levels of Caspase8 and Caspase 3 in ovaries from mice after fed with different diet for 20 weeks. (C) Intensities of cleaved-Caspase8 and cleaved-Caspase 3 normalized to β-actin. Data are presented as the mean ± standard deviation (n=3). # P<0.05 vs. 20W-NC group. NC: normal control diet group; HFD: high fat diet group; CL: corpus luteum; OC: oocyte; GC: granulosa cell; W: weeks.
DIO-type 2 diabetes suppressed ovarian angiogenesis
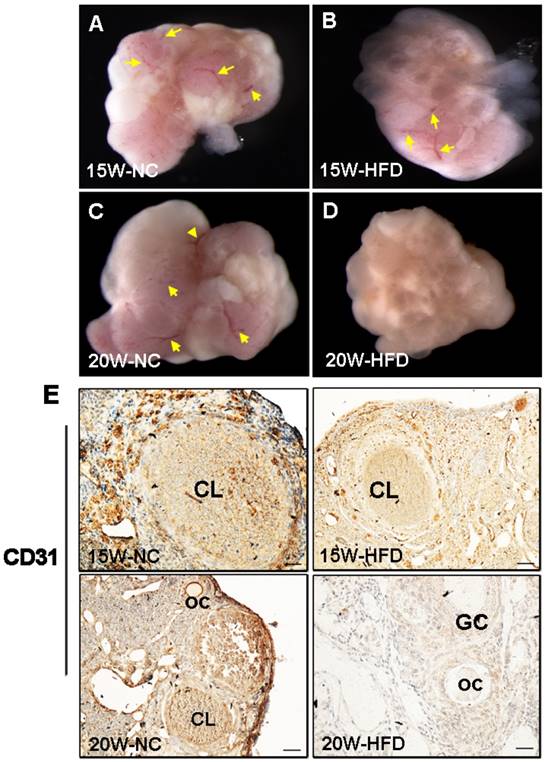
Angiogenesis is essential for normal ovarian function. In our present study, we found that the ovarian morphology of mice from 15W-HFD group shown normal ovarian morphology when compared with that in NC group with normal size and visible blood vessel (Figure 5A-C). By contrast, the ovaries of mice from 20W-HFD group exhibited hyaline appearance, much less blood vessel, and had large numbers of cystic follicles stored under the tunica albuginea (Figure 5D). Additionally, CD31 signals (the maker of vascular) of ovaries from 20W-HFD group were much week than those of ovaries from the other groups (Figure 5E).
DIO-type 2 diabetes suppressed angiogenesis of ovary. (A-D) Morphological appearance of ovaries from mice after 15/20 weeks fed with different mice. (E) Immunohistochemical staining of CD31 in ovaries from mice after 15/20 weeks fed with different diet, scale bars= 30 μm. Yellow arrow: blood vessel. NC: normal control diet group; HFD: high fat diet group; CL: corpus luteum; OC: oocyte; GC: granulosa cell; W: weeks.
DIO-type 2 diabetes inhibited HIF1α-VEGF signaling pathway in ovary
As a crucial transcription factor, HIF1α is involved in the regulation of VEGF expression and subsequent angiogenesis [20]. Here, we detected the expression of HIF1α and VEGF in ovary. It was observed that the mRNA levels of HIF1α and VEGF in ovaries from 20W-HFD group were significantly lower when compared with those in ovaries from 20W-NC group, but no significant difference between 15W-NC and 15W-HFD group (Figure 6A). Consistent with mRNA levels, diabetes significantly suppressed the protein levels of HIF1α and VEGF in ovaries from mice after fed with HFD for 20 weeks (Figure 6B). Immunohistochemical staining result had also shown much weaker signals of VEGF in ovary from 20W-HFD group (Figure 6C).
DIO-type 2 diabetes inhibited HIF1α-VEGF signaling pathway in ovary. (A) The mRNA levels of HIF1α and VEGF in ovaries from mice after 15/20 weeks fed with different diet. (B) The protein levels of HIF1α and VEGF in ovaries from mice after 15/20 weeks fed with different diet. (C-D) Immunohistochemical staining of VEGF in ovaries from mice after 15/20 weeks fed with different diet, scale bars= 30 μm. Data were presented as the mean ± standard deviation (n=3). *P<0.05 vs. 15W-NC group; #P<0.05 vs. 20 W NC groups. NC: normal control diet group; HFD: high fat diet group; CL: corpus luteum; OC: oocyte; GC: granulosa cell; W: weeks.

Discussion
Diabetes has a negative effect on ovarian follicular development and increases morbidity of infertility [25-29]. With the rapidly increasing epidemic of diabetes, it is an urgent need to further clarify the molecular mechanisms underlying diabetes-induced infertility. In our current study, we had successfully established type 2 diabetic mouse model in C57BL/6J mice via feeding with HFD to mimic human DIO-type 2 diabetes. After 15 weeks, the mice had not only been induced into obese, a major factor for induction of type 2 diabetes [30, 31], but also had the characteristics of type 2 diabetes. The mice were continuously fed with HFD and developed more serious characteristics of diabetes with higher body weight, blood glucose and more serious impaired glucose tolerance and insulin resistant after fed with HFD for 20 weeks.
Oocytes growth and GCs development are mutual and subtle regulatory processes during ovarian follicular development. Normal proliferation and apoptosis of GCs is essential for oocytes growth [32]. Follicular atresia occurs in the whole process of ovarian follicular development [33, 34]. GCs apoptosis is the main molecular mechanism of follicular atresia [35, 36]. Excessive apoptosis of GCs has a detrimental effect on ovarian follicular development. In current study, we found that apoptosis of GCs in COC, follicle wall and CL were significantly induced after 20 weeks on HFD. Our mechanistic study found that diabetes-induced excessive GCs apoptosis was Caspase 8-dependent, and finally activated Caspase 3.
Elevated cellular stress is a major causal event of diabetes-associated the onset of complications. Diabetes significantly induces oxidative stress [37], which in turn triggers DNA damage and ER stress [38], resulting in cell apoptosis [11, 39]. In present study, we found that diabetes significantly induced oxidative stress, up-regulated the expression of P53 gene, and activated the downstream molecular, CHK1 and CHK2, eventually caused DNA damage after fed with HFD for 20 weeks, but there are no significant difference in 15W-HFD group. Additionally, elevated ER stress was also involved in the process of diabetes-induced excessive apoptosis of GCs.
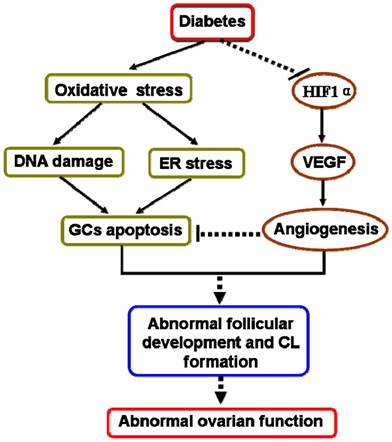
In our present study had also revealed that diabetes suppressed ovarian angiogenesis with much less blood vessels in ovary from mice in 20W-HFD group. We found that diabetes significantly suppressed HIF1α and subsequently inhibited the expression of VEGF, a vital regulatory factor for angiogenesis [40-42]. Angiogenesis is essential for normal follicular development, ovulation and CL formation [43, 44]. It is well known that comparing to the rest of developing follicles, dominant follicles have a higher degree of vascularity and thereby increase the supply of nutrients matching the demand from the maturing follicle [45]. Additionally, extensive vascular support is required for the phenotypic and functional shift of GCs [46]. These findings suggested that diabetes suppressed ovarian angiogenesis via inhibiting HIF1α-VEGF signaling pathway, which was one of potential molecular mechanisms for diabetes-induced abnormal ovarian function (Figure 7).
A schematic diagram illustrating the effect of diabetes on apoptosis of granulosa cells and ovarian angiogenesis in ovary. Type 2 diabetes not only induced oxidative stress, then triggered DNA damage and ER stress, thereby led to GCs apoptosis, but also inhibited HIF1α-VEGF signaling pathway and subsequently suppressed ovarian angiogenesis, consequently induced abnormal ovarian function.
Endothelial dysfunction is vital pathogenesis for diabetes-induced vascular complications, which is the principal cause of disability and death in patients with diabetes mellitus [47]. Reactive oxygen species (ROS) has been operated as signaling molecules to mediate various responses [48, 49], which involves in the modulation of HIF1α activity and VEGF expression. It was well known that hyperglycemia is the major risk factor for the development of diabetes-associated vasculopathy [50], which up-regulates the production of ROS in vascular cells [51, 52] and consequently mediates endothelial dysfunction [53]. However, the effect of hyperglycemia-induced elevated ROS on HIF1α is complicated. It has demonstrated that ROS, especially superoxide (O2-), suppresses HIF1α at the post-transcriptional level by activating a proline hydroxylase and finally ubiquitin-proteasome activity [54]. Additionally, ROS-NO and ROS-Rac1 signaling pathways are also the possible mechanisms underlying the impairment of the HIF1α pathway by hyperglycemia [54]. In our study, CD31, an endothelial marker, was significantly suppressed by high glucose, suggesting that hyperglycemia-induced elevated ROS maybe also involve in the suppression of HIF1α-VEGF signaling pathway and consequently lead to endothelial dysfunction and injury in ovary.
In our study, we chose to study the effect of diabetes on ovarian follicular development after fed with HFD for 15 weeks and 20 weeks. These findings had demonstrated that the effect of diabetes on ovarian function was time-dependent. The abnormal follicular development and suppression of angiogenesis were presented in ovaries from mice in 20W-HFD group. However, they weren't observed in ovaries from mice in 15W-HFD group. The mechanistic studies were consistent with these findings with the inhibition of HIF1α-VEGF signaling pathway, induction of DNA damage, and elevated ER stress in ovaries from 20W-HFD group. It is well known that prediabetes develops insulin resistance and triggers compensatory insulin secretion. Untreated, β-islet cells are destroyed after a long-term exposure to hyperglycemia, thereafter indispensable need for exogenous insulin supply [55, 56]. Consistent with prior study, we found that average blood glucose of mice from 20W-HFD group was significant higher than that of mice from 15W-HFD group, which may explain the adverse effect of diabetes on ovaries after fed with HFD for 20 weeks, but no remarkable influence at 15 weeks. Our results further confirmed that hyperglycemia resulting in glucotoxicity is the major factor that contributes to abnormal ovarian function in diabetes.
In summary, we used C57BL/6J mice to successfully establish DIO-type 2 diabetes. We found that diabetes not only significantly induced DNA damage and ER stress, thereby resulted in excessive GCs apoptosis, but also suppressed ovarian angiogenesis via inhibiting HIF1α-VEGF signaling pathway, leading to abnormal follicular development and CL formation in time-dependent (Figure 7). Hyperglycemia was the major pathogenic factor for diabetes-inducted abnormal ovarian function. These findings highlight that effective glucose control is necessary for normal ovarian function in diabetic mellitus.
Abbreviations
HFD: high fat diet; GCs: granulosa cells; CL: corpus luteum; ER: endoplasmic reticulum; VEGF: vascular endothelial growth factor; HIF1α: hypoxia inducible factor-1α; BMI: body mass index; GTT: glucose tolerance test; ITT: insulin tolerance test; H&E: hematoxylin and eosin; CHOP: C/EBP-homologous protein; DAB: 3, 3-diaminobenzidine; LPO: lipid hydroperoxide quantification; DIO: diet induced-obese; COC: cumulus-oocyte complex; p-CHK1: phosphorylated check point kinase1; p-PERK: phosphorylated protein kinase RNA-like ER kinase; p-IRE1α: phosphorylated inositolrequiring protein-1α; p-eIF2α: phosphorylated eukaryotic initiation factor 2α; CHOP: C/EBP-homologous protein; Bip: binding immunoglobulin protein.
Acknowledgements
This study was supported by a research grant from Zhejiang Provincial Natural Science Foundation of China (Y16H160214); Zhejiang Provincial Health department Key Scientific Project (2013RCA035); National Natural Science Foundation of China (31271255) and Fujian Provincial Natural Science Foundation (2016J01145).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gu L, Liu H, Gu X, Boots C, Moley KH, Wang Q. Metabolic control of oocyte development: linking maternal nutrition and reproductive outcomes. Cellular & Molecular Life Sciences. 2014;72:251-71
2. Moley NMG, Kelle H. Maternal obesity, infertility and mitochondrial dysfunction: potential mechanisms emerging from mouse model systems. Molecular human reproduction. 2013;19:486-94
3. Chang AS, Dale AN, Moley KH. Maternal diabetes adversely affects preovulatory oocyte maturation, development, and granulosa cell apoptosis. Endocrinology. 2005;146:2445
4. Wang Q, Chi MM, Schedl T, Moley KH. An intercellular pathway for glucose transport into mouse oocytes. Ajp Endocrinology & Metabolism. 2012;302:E1511-8
5. Alonso-Pozos I, Rosales-Torres AM, Avalos-Rodríguez A, Vergara-Onofre M, Rosado-García A. Mechanism of granulosa cell death during follicular atresia depends on follicular size. Theriogenology. 2003;60:1071-81
6. Palumbo A, Yeh J. Apoptosis as a basic mechanism in the ovarian cycle: follicular atresia and luteal regression. Reproductive Sciences. 1995;2:565
7. Wu Y, Zhang Z, Liao X, Wang Z. High Fat Diet Triggers Cell Cycle Arrest and Excessive Apoptosis of Granulosa Cells during the Follicular Development. Biochemical & Biophysical Research Communications. 2015;466:599-605
8. Wu Y, Wang F, Fu M, Wang C, Quon MJ, Yang P. Cellular stress, excessive apoptosis and the effect of metformin in a mouse model of type 2 diabetic embryopathy. Diabetes. 2015;64:2526-36
9. Wu Y, Wang F, Reece EA, Yang P. Curcumin ameliorates high glucose-induced neural tube defects by suppressing cellular stress and apoptosis. American Journal of Obstetrics & Gynecology. 2015;212:1-8
10. Wu Y, Reece EA, Zhong J, Dong D, Shen WB, Harman CR. et al. Type 2 diabetes mellitus induces congenital heart defects in murine embryos by increasing oxidative stress, endoplasmic reticulum stress and apoptosis. American Journal of Obstetrics & Gynecology. 2016;215:366.e1-e10
11. Grindel A, Guggenberger B, Eichberger L, Pöppelmeyer C, Gschaider M, Tosevska A. et al. Oxidative Stress, DNA Damage and DNA Repair in Female Patients with Diabetes Mellitus Type 2. PloS one. 2016;11:e0162082
12. Hamdan M, Jones KT, Cheong Y, Lane SI. The sensitivity of the DNA damage checkpoint prevents oocyte maturation in endometriosis. Scientific reports. 2016;6:36994
13. Tamanini C, De AM. Angiogenesis in developing follicle and corpus luteum. Reproduction in Domestic Animals. 2004;39:206
14. Robinson RS, Woad KJ, Hammond AJ, Laird M, Hunter MG, Mann GE. Angiogenesis and vascular function in the ovary. Reproduction. 2009;138:869-81
15. Wulff C, Wilson H, Wiegand SJ, Rudge JS, Fraser HM. Prevention of thecal angiogenesis, antral follicular growth, and ovulation in the primate by treatment with vascular endothelial growth factor Trap R1R2. Endocrinology. 2002Jul;143(7):2797-807
16. Zimmermann RC, Hartman T, Kavic S, Pauli SA, Bohlen P, Sauer MV. et al. Vascular endothelial growth factor receptor 2-mediated angiogenesis is essential for gonadotropin-dependent follicle development. Journal of Clinical Investigation. 2011;112:659
17. Qiu Y, Seager M, Osman A, Castlemiller J, Bevan H, Tortonese DJ. et al. Ovarian VEGF(165)b expression regulates follicular development, corpus luteum function and fertility. Reproduction. 2012;143:501-11
18. Fu Y, Lai Y, Wang Q, Liu X, He W, Zhang H. et al. Overexpression of clusterin promotes angiogenesis via the vascular endothelial growth factor in primary ovarian cancer. Molecular medicine reports. 2013;7:1726
19. Pan XY, Zhang ZH, Wu LX, Wang ZC. Effect of HIF-1a/VEGF signaling pathway on plasma progesterone and ovarian prostaglandin F2a secretion during luteal development of pseudopregnant rats. Genetics & Molecular Research Gmr. 2015;14:8796-809
20. Park ST, Kim BR, Park SH, Lee JH, Lee EJ, Lee SH. et al. Suppression of VEGF expression through interruption of the HIF-1α and Akt signaling cascade modulates the anti-angiogenic activity of DAPK in ovarian carcinoma cells. Oncology reports. 2014;31:1021
21. ZH Z, LY C, F W, YQ W, JQ S, XH H. et al. Expression of hypoxia-inducible factor-1α during ovarian follicular growth and development in Sprague-Dawley rats. Genetics and molecular research: GMR. 2015;14:5896-909
22. Zhang B, Wang D, Ji TF, Shi L, Yu JL. Overexpression of lncRNA ANRIL up-regulates VEGF expression and promotes angiogenesis of diabetes mellitus combined with cerebral infarction by activating NF-κB signaling pathway in a rat model. Oncotarget. 2017
23. Zheng P, Lin Y, Wang F, Luo R, Zhang T, Hu S. et al. 4-PBA Improves Lithium-induced Nephrogenic Diabetes Insipidus by Attenuating ER Stress. American Journal of Physiology Renal Physiology. 2016;311(4):F763
24. Wang F, Wu Y, Quon MJ, Li X, Yang P. ASK1 mediates the teratogenicity of diabetes in the developing heart by inducing ER stress and inhibiting critical factors essential for cardiac development. American journal of physiology Endocrinology and metabolism. 2015;309:487-99
25. Tavakolian AA, Nouri M, Tavakolian AS. Surprisingly low infertility rate in married type 2 diabetic women: A rather curious paradox to the current opinion of insulin resistance as the joint pathogenesis of poly cystic ovary syndrome and type 2 diabetes mellitus. Diabetes & Metabolic Syndrome Clinical Research & Reviews. 2015;9:201-4
26. Nandi A, Poretsky L. Diabetes and the female reproductive system. Endocrinology & Metabolism Clinics of North America. 2013;42:915-46
27. Gu L, Liu H, Gu X, Boots C, Moley KH, Wang Q. Metabolic control of oocyte development: linking maternal nutrition and reproductive outcomes. Cellular & Molecular Life Sciences. 2015;72:251-71
28. Colton SA, Pieper GM, Downs SM. Altered Meiotic Regulation in Oocytes from Diabetic Mice. Biology of Reproduction. 2002;67:220-31
29. Maheshwari A, Stofberg L, Bhattacharya S. Effect of overweight and obesity on assisted reproductive technology—a systematic review. Human Reproduction Update. 2007;13:433-44
30. King AJ. The use of animal models in diabetes research. British journal of pharmacology. 2012;166:877-94
31. Kahn SE, Hull RL, Utzschneider KM. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2007;444:840-6
32. Li Q, Mckenzie LJ, Matzuk MM. Revisiting oocyte-somatic cell interactions: in search of novel intrafollicular predictors and regulators of oocyte developmental competence. Mol Hum Reprod. 2008Dec;14(12):673-8
33. Hirshfield AN. Development of follicles in the mammalian ovary. International Review of Cytology. 1991;124:43-101
34. Matsuda F, Inoue N, Manabe N, Ohkura S. Follicular growth and atresia in mammalian ovaries: regulation by survival and death of granulosa cells. Journal of Reproduction & Development. 2012;58:44
35. Palumbo A, Yeh J. Apoptosis as a basic mechanism in the ovarian cycle: follicular atresia and luteal regression. Reproductive Sciences. 1995;2:565-73
36. Alonso-Pozos I, Rosales-Torres AM, Ávalos-Rodrı́Guez A, Vergara-Onofre M, Rosado-Garcı́A A. Mechanism of granulosa cell death during follicular atresia depends on follicular size. Theriogenology. 2003;60:1071-81
37. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813-20
38. Wang F, Reece EA, Yang P. Superoxide dismutase 1 overexpression in mice abolishes maternal diabetes—induced endoplasmic reticulum stress in diabetic embryopathy. American Journal of Obstetrics & Gynecology. 2013;209:1-7
39. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. Faseb Journal Official Publication of the Federation of American Societies for Experimental Biology. 2003;17:1195
40. Abe K, Shoji M, Chen J, Bierhaus A, Danave I, Micko C. et al. Regulation of vascular endothelial growth factor production and angiogenesis by the cytoplasmic tail of tissue factor. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:8663-8
41. Zhang Z, Neiva KG, Lingen MW, Ellis LM, Nör JE. VEGF-dependent tumor angiogenesis requires inverse and reciprocal regulation of VEGFR1 and VEGFR2. Cell Death & Differentiation. 2009;17:499
42. Ferrara N. Vascular endothelial growth factor and the regulation of angiogenesis. Recent Progress in Hormone Research. 2000;55:15-35
43. Abulafia O, Sherer DM. Angiogenesis of the ovary. American Journal of Obstetrics & Gynecology. 2000;182:240-6
44. Redmer DA, Reynolds LP. Angiogenesis in the ovary. Reviews of Reproduction. 1996;1:182-92
45. Zeleznik AJ, Schuler HM Jr RL. Gonadotropin-binding sites in the rhesus monkey ovary: role of the vasculature in the selective distribution of human chorionic gonadotropin to the preovulatory follicle. Endocrinology. 1981;109:356
46. Ramakrishnan S, Subramanian IV, Yokoyama Y, Geller M. Angiogenesis in normal and neoplastic ovaries. Angiogenesis. 2005;8:169
47. Potenza MA, Gagliardi S, Nacci C, Carratu' MR, Montagnani M. Endothelial dysfunction in diabetes: from mechanisms to therapeutic targets. Current Medicinal Chemistry. 2009;16:94-112
48. Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882
49. Forman HJ, Maiorino M, Ursini F. Signaling functions of reactive oxygen species. Biochemistry. 2010;49:835-42
50. Cade WT. Diabetes-Related Microvascular and Macrovascular Diseases in the Physical Therapy Setting. Physical Therapy. 2008;88:1322
51. Batchuluun B, Inoguchi T, Sonoda N, Sasaki S, Inoue T, Fujimura Y. et al. Metformin and liraglutide ameliorate high glucose-induced oxidative stress via inhibition of PKC-NAD(P)H oxidase pathway in human aortic endothelial cells. Atherosclerosis. 2014;232:156-64
52. Yu T, Jhun BS, Yoon Y. High-Glucose Stimulation Increases Reactive Oxygen Species Production Through the Calcium and Mitogen-Activated Protein Kinase-Mediated Activation of Mitochondrial Fission. Antioxidants & Redox Signaling. 2011;14:425
53. Yang C, Lei W, Pitzer AL, Xiang L, Li PL, Yang Z. Contribution of redox-dependent activation of endothelial Nlrp3 inflammasomes to hyperglycemia-induced endothelial dysfunction. Journal of Molecular Medicine. 2016:1-13
54. Xiao H, Gu Z, Wang G, Zhao T. The Possible Mechanisms Underlying the Impairment of HIF-1α Pathway Signaling in Hyperglycemia and the Beneficial Effects of Certain Therapies. International Journal of Medical Sciences. 2013;10:1412-21
55. Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. Journal of Clinical Investigation. 2006;116:1802
56. Kasuga M. Insulin resistance and pancreatic β cell failure. Journal of Clinical Investigation. 2006;116:1756-60
Author contact
![]() Corresponding authors: Yanqing Wu, yqwu220946net Jian Xiao, xfxj2000com
Corresponding authors: Yanqing Wu, yqwu220946net Jian Xiao, xfxj2000com